
INTRODUCTION
A microarray is a robust tool in molecular research used in studying gene expression for the whole genome under specific conditions (Seder et al., 2021). Ribonucleic acid (RNA) is an essential polymeric molecule found in biological cells which plays a vital role in the coding, decoding, regulation, and expression of cellular genes. RNA exists in several forms, including ribosomal RNA (80%–90%), messenger RNA (2.5%–5%), and transfer RNA (Sealfon and Chu, 2011). RNA profiling studies are extremely essential in elucidating the function of Messenger RNA (mRNA) in regulating protein synthesis, cellular cycles, apoptosis, and the differential regulation of biological pathways (Pusic et al., 2021). Expression analysis for the functional mRNA is ultimately required to provide a comprehensive understanding of both cellular networking and cellular regulatory mechanisms.
mRNA conveys the genetic information to retain life through mediating protein synthesis (Guttman et al., 2021). Furthermore, the alterations in RNA expression levels in diseases make them a hallmark for diagnostic and prognostic purposes and drug binding sites in the pharmaceutical industry. Recently, mRNA has been implemented as a vaccine in the COVID-19 pandemic (Dolgin, 2021).
Therefore, several platforms have been developed to quantify mRNA and to analyze RNA expression, such as deoxyribonucleic acid (DNA) microarray, northern blotting, and real-time quantitative reverse transcription PCR (RT-qPCR) (Ngassam Tchamba et al., 2019). Furthermore, more reliable and affordable techniques such as RNA sequencing using a next-digital reverse transcription polymerase chain reaction (RT-PCR) and generation DNA sequencer were developed to study mRNA expression levels in detail (Le Rhun et al., 2016). The microarray and RT-qPCR detection methods are highly dependent on the quality of the RNA extract to provide reliable and precise results (Sealfon and Chu, 2011). It is becoming evident that there is a crucial need to optimize and standardize the isolation of RNA, given the discrepancies among many studies, whereby the recovery of RNA is dependent on the extraction method used.
RNA extraction relies strongly on good laboratory practice and a nuclease-free environment (Buckingham, 2019). Extraction methods could be divided according to the protocol into two major methods: the usage of 4M guanidinium thiocyanate and the usage of phenol SDS. More recently, the usage of magnetic beads has been implemented in RNA extraction procedures (Rodríguez et al., 2020). RNA extraction includes several steps such as cellular disruption, protein precipitation, and nucleic acid purification. The cellular disruption is performed through three main processes: first, the robust mechanical forces using glass or magnetic beads, secondly, using a chaotropic agent, and, eventually, utilizing enzymatic cellular digestion (He et al., 2017). Protein precipitation is performed through adding high concentrations of salt or even changing the pH of the buffer. RNA extraction is carried out by the conventional liquid-liquid extraction method or the commercially available solid extraction method (Metcalf and Weese, 2012). Each method has advantages and discrepancies over the other method. For instance, liquid-liquid extraction is characterized by feasibility and abundance of constituencies in research laboratories. On the other hand, this method lacks specificity and high output quality (Bustin et al., 2009), whereas solid-phase extraction is characterized by simplicity and specificity but regrettably a high cost. Substantially, several scientists reported that their discrepancies in gene expression levels are regarded as variations in RNA isolation techniques and not as alterations in gene expression levels (Heera et al., 2015; Rodríguez and Vaneechoutte, 2019; Rodríguez et al., 2020). Regrettably, many studies lack proper RNA quality control, such as RNA integrity (Brunet-Vega et al., 2015).
No doubt, for conducting a high-throughput RNA analysis, niche RNA quality is an imperative (Mommaerts et al., 2015). Unfortunately, the techniques used for RNA analysis have been developed rapidly, providing higher sensitivity and specificity, whereas the techniques used for RNA extraction did not develop consequently in the same pattern.
A series of precautions and special care during RNA extraction are required since RNA is highly susceptible to degradation by the endogenous nucleases ubiquitously present in both blood tissue and most bacteria (Bayatti et al., 2014). Inadequate RNA extraction hampers fundamental information about gene expression and cellular regulation, which means RNA is not reflecting absolute accurate levels (Wong et al., 2019). Before conducting RNA extraction, appropriate sample storage is required since RNA is vulnerable to degradation by RNAs, which substantially affects the gene expression levels. Moreover, transcription and translation can continue even after sample collection, so RNA levels present during the analysis process are not reflecting the RNAs at the time of collection (Pusic et al., 2021). The process of submerging the collected samples in a cryogenic solution (-180°C) can ultimately stop the expression levels in the biological cells. Unfortunately, the cryogenic technique is not available to all scientists in many researches due to safety reasons and lacking infrastructure. The development of a simple, accurate, robust method for RNA extraction is mandatory to increase reproducibility, decrease the feasibility, and produce a high throughput (Yip et al., 2017). Thus far, there is a dearth in the number of studies that compared the extraction of RNA from biofilms in a methodological manner (Liu et al., 2015).
A biofilm is one of the forms of microorganisms commonly found in nature, environmental systems, and the industrial and medical fields. Biofilms are generally known as communities of microbes that are attached to certain surfaces that are normally covered with an extracellular matrix (ECM) secreted by the same microbes (Kim et al., 2021). ECM is constituted of exopolysaccharide, extracellular DNA, RNA, proteins, and lipids. Bacteria in biofilms are more resistant to environmental factors leading to chronic infections in the host (Wei and Ma, 2013). Pseudomonas aeruginosa (P. aeruginosa) and Streptococcus pyogenes (S. pyogenes) are considered a major cause of human acquired infection owing to their ability to form biofilms, the conversion from the planktonic to the biofilm stage, and change the gene expression pattern, which contributes to antibiotic resistance enhancement (Thi et al., 2020).
In this study, we will evaluate the quantity and integrity of RNA extracted from two bacterial biofilms, S. pyogenes [American type culture collection (ATCC) 19615] and P. aeruginosa (ATCC 10145), using three common RNA extraction kits [Spin or vacuum (SV) Total RNA Isolation System, RNeasy Mini Kit, and TRIzol Reagent]. Furthermore, we will describe the modifications we applied in the extraction process to achieve both higher RNA throughput and integrity numbers.
MATERIALS AND METHODS
Bacterial culture
Initially, two bacterial strains were utilized in the current study: S. pyogenes (ATCC 19615) and P. aeruginosa (ATCC 10145). The bacteria were cultured in 20 ml of Tryptic Soya Broth (TSB) (Fisher Scientific, UK) in a shaker incubator (150 rpm) at 37ºC for 24 hours under aerobic conditions (Seder et al., 2021).
Viable bacterial count
To attain the appropriate bacterial count to standardize the biofilm experiments, an overnight culture was incubated at 37ºC for 16 hours in a nutrient broth in an incubator-shaker with the rotation speed of 150 rpm. After incubation, a series of serial dilutions was conducted for the bacterial culture in a 10-fold dilution until the dilution 10−4 was reached. The optical density (OD) at wavelength 600 nm was measured for each dilution using a spectrophotometer (Eppendorf, USA), and the readings were recorded for further steps. Later, 100 μl of each serial dilution was suspended on a nutrient agar and spread using a dispenser. Plates were incubated at 37ºC for 16 hours, and the colonies of each plate were counted individually. The OD of the appropriate dilution was used for the biofilm formation assay. Eventually, the final count of the viable bacteria was set to be approximately around 1.5×108 colony-forming units (CFUs/ml) for the two bacterial strains (Mirani et al., 2018).
Biofilm formation
The biofilm formation assay was performed using a tissue culture plate (Hassan et al., 2011), and the strength of biofilm formation was determined using the crystal violet method (Fisher Scientific, UK).
Nunc plates of 12 wells were used to establish bacterial biofilms by dispensing 2 ml of each bacterial suspension in TSB fortified with 1% glucose (w/v) and incubated at 37ºC for 24 hours. Later, the planktonic growth around the biofilms was aspirated carefully and washed three times using phosphate buffer saline (PBS) (Invitrogen, UK). After that, the plates were air-dried by being inverting in a twisted position for 15 minutes at 25ºC. The wells were stained using 2 ml of 0.1% (w/v) crystal violet and incubated at 25ºC for 15 minutes. The excessive amounts of crystal violet were removed by washing the plates three times with PBS. Eventually, the strength of biofilm formation was quantified by measuring the dye deduced from the wells by pipetting 2 ml of 95% ethanol on biofilms, and the absorbance was determined at 570 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader (Tecan Infinite 200 PRO, Austria). The experiment was conducted in triplicate, and the average and standard deviation was calculated (Hassan et al., 2011).
Biofilm degradation assay
Biofilms were established as mentioned previously, and later, the supernatant was removed by pipetting and replaced with 2 ml of 20% (w/v) Trigona honey that was diluted in TSB supplemented with 1% glucose (w/v). The plates were further incubated at 37°C for 2 hours. TSB + 1% glucose was used as a negative control, whereas bacterial suspension without honey was used as a growth control. After the incubation, the planktonic growth around the biofilms was aspirated carefully and washed three times using PBS (Invitrogen, UK). After that, the plates were air-dried by being inverting in a twisted position for 15 minutes at 25ºC. The wells were stained and measured as mentioned in biofilm formation, and the absorbance was determined at 570 nm using an ELISA plate reader (Tecan Infinite 200 PRO, Austria). The experiment was conducted in triplicate, and the average and standard deviation were calculated (Maleševi? et al., 2019).
RNA extraction
To extract RNA from the established bacterial biofilms, P. aeruginosa and S. pyogenes were cultivated as mentioned previously in biofilm formation. After incubation, the plates were placed on ice, and the planktonic growth above the biofilms was aspirated carefully using a micropipette. Later, the biofilms were scraped and suspended into 1 ml sterile distilled water and then vortexed for 1 minute to break up cell aggregates before further analysis was conducted. RNA was extracted using three different kits, and the procedures were conducted according to the manufacturer’s instructions. 1) RNeasy Mini Kit System (Promega, UK): 1 ml of the biofilm was suspended in a buffer solution until OD600 = 0.6 was reached. Cell wall digestion was conducted using a mixture of 60 μl of 10 mg/ml lysozyme and 60 μl of 10 mg/ml lysostaphin for 20 minutes at 25ºC. Later, 75 μl of RNA lysis buffer was added, and the process was continued per the manufacturer’s instructions. Eventually, 100 μl of nuclease-free water was used for RNA elution using RNA extraction MicroSpin Columns (Beltrame et al., 2015). 2) RNeasy Mini Kit (QIAGEN, Hilden, Germany): 1 ml of biofilm suspension was treated with 100 μl of 0.1 mg/ml lysosome (Sigma-Aldrich, Gillingham, UK) for 30 minutes at 37ºC, and RNA was extracted from 400 μl of digested sample and eluted in 50 μl of RNase-free water using RNA extraction MicroSpin Columns (Atshan et al., 2012). 3) TRIzol Reagent (Thermo Fisher Scientific): 100 μl of the suspended biofilm was dissolved in 900 μl of TRIzol Reagent, mixed, and vortexed, then the procedure was performed according to the company leaflet, and lastly, RNA was precipitated using 100% ethanol and eventually RNA was suspended in 50 μl of RNase-free water. The modified method of RNA extraction relied on using a mixture of the three procedures. Firstly, enzymatic digestion was applied using 100 μl of 0.1 mg/ml lysosome with the biofilm aspirate (Sigma-Aldrich, Gillingham, UK) for 30 minutes at 37ºC. Later, TRIzol Reagent was applied to perform liquid-liquid extraction for RNA. 100 μl of the lysate digestion was mixed and vortexed with 900 μl of TRIzol Reagent and vortexed vigorously. Lastly, MicroSpin® Columns (Promega, UK) were used to conduct mRNA extraction. The total amount of the aqueous phase of TRIzol Reagent was pipetted directly into MicroSpin Columns, and the procedure was carried out as per the instructions from the Promega company. RNase-free water was used as a negative control in all experiments as internal control, and RNA samples were stored at -80ºC for further investigation (Cury and Koo, 2007).
RNA quality control
Total RNA concentration and purity were determined using NanoDrop (Implen, Germany). According to the manufacturer’s instructions, 1 μl of RNA extract was directly applied to the optic lens. The purity of the RNA was examined using the 260/230 and 260/280 absorbance ratios. Samples with ratios between 1.8 and 2.1 were accepted for further analysis.
Preliminarily, RNA integrity was examined visually using gel electrophoresis. 1.5% (w/v) of agarose (Bio-Rad, USA) powder was suspended in 0.5 × TBE buffer, and the mixture was subjected to a microwave oven for 3 minutes until boiling was reached. Later, the mixture was poured into a gel tray of the electrophoresis apparatus containing combs and allowed to sit for 20 minutes. 1 μl of RNA extract was pipetted on the agarose gel, and electrophoresis was settled at 80 V for 1 hour. The gel was soaked in a solution containing 0.5 μg/ml of SYBR™ Green for 20 minutes. The gel was visualized using the Biometra system, and the images were stored on disks as tag image file format files. The gel was examined for the presence of the ribosomal 16s and the 23s bands of both P. aeruginosa and S. pyogenes. Eventually, the RNA samples which matched the completion criterion were subjected to the 2100 Agilent Bioanalyzer to determine the RNA integrity number (RIN). All samples were stored at -80ºC for further analysis (Rayyan et al., 2019) (the experiment was performed in duplicate).
Quantitative real-time PCR
The cDNA library was accomplished by converting total RNA samples to cDNA using the GoTaq® 2-Step RT-qPCR System (Promega, Southampton, UK). A master mixture of 0.5 μg random primers, 0.5 μg oligo (dT) primer, 0.5 μg of total RNA, and nuclease-free water at a whole volume of 10 μl was prepared, settled at 70ºC for 5 minutes using a thermal cycler (AB Applied Biosystems). After that, samples were chilled on ice for 5 minutes, and 1 μl of the mixture was used in the complementary DNA synthesis. According to the vendor’s instructions, a master mix of 1 μl PCR Nucleotide Mix, 4 μl of 5× GoScript RT Buffer, 1 μl GoScript™ Reverse Transcriptase, 2 μl of MgCl2 was prepared, and the total volume was filled with nuclease-free water to 20 μl. The incubation process of the mixture was accomplished using a Bio-Rad thermal cycler at 42ºC heat block for 1 hour, and then further incubation was conducted at 72ºC for 15 minutes for enzyme inactivation (Seder et al., 2021).
The primers used for RT-PCR analysis are listed in Table 1. The RT-PCR reaction was conducted according to the company’s instructions. A master mix of 20 μl was prepared by mixing 1 μl of 10 PM of forwarding primer, 1 μl of 10 PM of reverse primer, 2 μl of cDNA template, and 10 μl of PCR Master Mix and topped up with nuclease-free water to 20 μl. The PCR protocol of Rotor-Gene Q (QIAGEN, Hilden, Germany) was used for the amplification and determination process. Three biological samples were used as replicates in performing the analysis.
RESULTS
Viable bacterial count
To start with an accurate bacterial number in the biofilm experiments, we performed the (CFUs/ml) assay for both P. aeruginosa (ATCC 10145) and S. pyogenes (ATCC 19615) after incubation for 24 hours at 37ºC, and the CFU number was counted.
In reference to the comparison in Figure 1, a clear difference between CFU values for both bacterial strains was noticed when we conducted the CFU assay based on the McFarland standard. Therefore, to start our experiments with the same bacterial count, we adopted a viable CFU count instead of absolute OD values, and we calculated the relevant OD value which will yield a number of around 1.5×108 bacteria; therefore, we prepared a standard curve using the McFarland standards 0.5–4, and we found that OD600 = 0.25 for P. aeruginosa produces 1.5×108 CFUs/ml, while OD600 = 0.65 for S. pyogenes produces 1.5×108 CFUs/ml.
Biofsilm formation assay
The biofilm formation assay was used to determine the strength of the biofilms formed by P. aeruginosa (ATCC 10145) and S. pyogenes (ATCC 19615). The biofilms were grown for 24 hour before being measured and compared with the negative control to classify the strength of adherence. Both bacterial strains have developed biofilms. The OD600 of the biofilms formed by both strains was four times higher than the OD600 of the negative control, so, according to the interpretation guidelines (Cerca et al., 2005), biofilms formed by P. aeruginosa and S. pyogenes are classified as strong adherent biofilms.
Biofilm degradation assay
To study the impact of applying Trigona honey on established biofilms of P. aeruginosa (ATCC 10145) and S. pyogenes (ATCC 19615), biofilms were established as mentioned previously, and then honey was added at a concentration of 20% (w/v) for 2-hour treatment. A relevant 45.6% (p < 0.001) reduction in P. aeruginosa biomass was achieved, while a 61.9% (p < 0.001) reduction in S. pyogenes biomass was noticed. Trigona honey showed a significant biofilm degradation activity at a concentration of 20% (Fig. 2).
RNA extraction and quality control
The scientific committee recommends several critical tests for quality assurance of RNA to test RNA quality and integrity before proceeding with RNA interrogation through a molecular tool such as RNA sequencing or microarray analysis or even RT-PCR. In the current study, the ratios of 260/280 for extracted RNA from different biofilms were between 1.8 and 2.1, whereas the RNA integrity using the three commercial kits was low RIN between 2.3 and 4.1 (Fig. 3, Table 2).
Meanwhile, the extracted RNA samples in the procedure of the modified method matched the criteria (No. 4, Table 2) as RNA was intact on agarose gel electrophoresis (Fig. 3), and RNA integrity ranged from 7.9 to 9 for P. aeruginosa and S. pyogenes when analyzed using the 2100 Agilent Bioanalyzer (Table 2, Fig. 4 and 5). The outcome of extracted RNA has been exposed to a wide variation in final extract amount as the concentrations ranged between 90 and 465 ng/ml using the three commercial kits, whereas the RNA outcome reached 1,321 ng/ml using modified method No. 4 (Table 3).
DISCUSSION
For this study, we quantified the mRNA extracted from two bacterial strains, P. aeruginosa and S. pyogenes, in the biofilm state. No doubt, RNA yield and integrity are indispensable factors denoting the quality of the downstream molecular analysis tools. However, RNA quality is considered as a criterion for evaluating the quality of the extraction process. Nonetheless, as scientists, we ubiquitously face the aberrant results of combined RNA low yield and high disintegration. The low output could be attributed to distinct factors, among them the inappropriate extraction procedure or a low starting number of cells or even the cellular disintegration caused by environmental or RNAs activity (Kashofer et al., 2013). The accuracy and precision of gene expression are substantially pertinent to RNA quality (Watermann et al., 2016). Therefore, the way of sample handling during the experiment performance, the procedure of RNA extraction, and the kit of choice to extract RNA are drastically crucial (Bayatti et al., 2014; Mack et al., 2007).
 | Table 1. Primers for RT-PCR. [Click here to view] |
 | Figure 1. Colony-forming units for P. aeruginosa and S. pyogenes at several OD600 values. [Click here to view] |
 | Figure 2. Comparison of the % degradation of 24 hours biofilm for P. aeruginosa and S. pyogenes. There was significant degradation in the biofilm mass relevant to the 20% (w/v) of Trigona honey showing deterioration of 45.6% of P. aeruginosa biofilm mass, while there was 61.9% biofilm degradation for S. pyogenes. *** = p < 0.001. [Click here to view] |
Several microbiologists recommend the McFarland standard in the harvesting of the bacterial cultures and starting inoculation numbers for bacterial cultures or even for adjusting the bacterial number for proper RNA extraction (Kafantaris et al., 2021; Romero et al., 2018; Sonnleitner et al., 2017). We showed there is a significant variation in bacterial number between P. aeruginosa and S. pyogenes when we adjusted the inoculum to 0.6 Abs at OD600 as recommended in RNA extraction kits. Therefore, based on the current results, the usage of viable bacterial count is considered more precise and accurate than other spectrophotometer methods. Likewise, Peñuelas-Urquides et al. (2013) showed a variation in the viable bacterial number of Mycobacterium tuberculosis and the values proposed by the McFarland standard.
In this study, three RNA extraction kits (SV Total RNA Isolation System, RNeasy Mini Kit, and TRIzol LS) were utilized to extract RNA from two opportunistic bacterial strains, P. aeruginosa and S. pyogenes, in the biofilm state under the conditions of the presence and absence of Trigona honey. RNA purity was assessed through NanoDrop, integrity through 2100 Bioanalyzer, and quantity through NanoDrop, 2100 Bioanalyzer, and RT-qPCR. The results obtained from the first three commercial kits showed low RNA yield and low integrity numbers when RNA was measured using NanoDrop and 2100 Bioanalyzer. The outcome of RT-PCR was not influenced by RNA integrity since the RIN values of more than two are considered as a criterion suitable for the RT-PCR test. On the contrary, the RNA yield influences the RT-PCR result reciprocally as the higher the yield of RNA, the less the cycle threshold (ct) value (Bustin et al., 2009). Regrettably, RNA extraction results did not match the quality criterion, namely, the integrity number, regardless of the extraction method utilized in the three extraction kits; therefore, some modifications were made in the extraction procedure to accomplish high RNA yield with a low disintegration ratio. To rule out personal error, the experiments were performed in triplicate, and there was no bias toward any company over the others. On the contrary, we applied the extraction procedure according to the vendor’s instructions precisely. The elusive and sensitive nature of RNA caused distinct problems and threats during the extraction process, among them RNA degradation by environmental RNAs or contamination with proteins and salts. It is required to reach a converge point by which RNA could be extracted in high yield and decent integrity texture. RNA extraction is simply a confounder as the process could be summarized by several steps, and then the targeted RNA will be collected.
Herewith, we report that not only was the quality of RNA improved but also the efficiency of RNA extraction was implicated when we used a mixture of liquid-liquid extraction and solid-phase extraction in RNA extraction. The first obstacle to extracting an adequate yield of RNA with high integrity is the digestion of bacterial cell walls. According to the vendors’ instructions, the cell wall of bacteria was subjected to enzymatic digestion using lysozyme and lysostaphin, then followed by spheroplast lysis using the lysis buffer for the SV Total RNA Isolation System and RNeasy Mini Kits, whereas the phenol activity of the TRIzol LS kit (Sambrook et al., 1989) was added directly without adding further enzymes. Regrettably, this procedure did not confer a high output of RNA for any commercial kit even though we applied each step precisely. On the contrary, the amounts of RNA did not reach 100 ng/μl with a low integrity number less than four. Therefore, a congestive plan was designed to manipulate the procedures and to deduce the strong points from each kit. We decided to start with enzymatic digestion followed by liquid-liquid extraction using the TRIzol kit and eventually applying the MicroSpin Columns of both the SV Total RNA Isolation System and RNeasy Mini Kit. The outcomes were exceptionally good and matched the requirements. Considerable amounts of RNA were extracted with high yield and integrity numbers. The RNA output reached 1,300 ng/μl, and the integrity number started from 7 to 9.8. These values are highly reliable for downstream molecular analysis such as microarray or RNA sequencing.
To determine the quality of RNA, recommended measurement A260/A230 and A260/A280 ratios are adopted (Manchester, 1996; Sambrook et al., 1989). Contamination by organic solvents or TE buffer is determined by the absorbance at 230 nm, whereas nucleic acids are measured at absorbance of 260 nm, and protein contamination is measured by absorbance at 280 nm. The measurement of the A260/A280 ratio is an imperative step to interoperate the contamination level, which in turn could inhibit the reaction of enzymes, i.e., reverse transcriptase and DNA polymerase (Zhong et al., 2020). The A260/A280 and A260/A230 ratios are widely accepted for evaluating the contamination in the prepared RNA samples. Even though these ratios do not directly evaluate the inhibition of enzyme activity, they are a criterion for downstream investigations (Tavares et al., 2011). RNA purity is commonly evaluated by measuring the ratio between the absorbance at 260 nm and 280 nm (A260/A280) absorbance, whereby a value of ~2.0 is generally accepted as indicating that the RNA is free of proteins. Extracted RNA samples are partially incorporated with contaminants such as proteins, polysaccharides, and even salts. These contaminants can interfere with RNA interrogation in unwanted enzymatic reactions that may inhibit RNA extraction and, hence, denote false-negative results (Pionzio and McCord, 2014). RNA integrity could be evaluated using capillary gel electrophoresis with the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) or even manually by running gel electrophoresis using agarose gel followed by RNA staining with ethidium bromide or SYBR™ Green (Grobe et al., 2019). The Agilent method calculates the RNA integrity number (RIN) value by comparing the 18S/28S ribosomal ratio to determine mRNA quality (Imbeaud et al., 2005). Even though the Agilent method is considered precise and reliable, regrettably, the instrument is not available in all research institutes, whereas gel electrophoresis can be performed in most research institutes in regard to feasibility and affordability. The drawback of gel electrophoresis is that no number will be generated after the evaluation to endow a definitive decision on the RNA quality. Rather, the process is an optical evaluation of the integrity, and no disintegration is present.
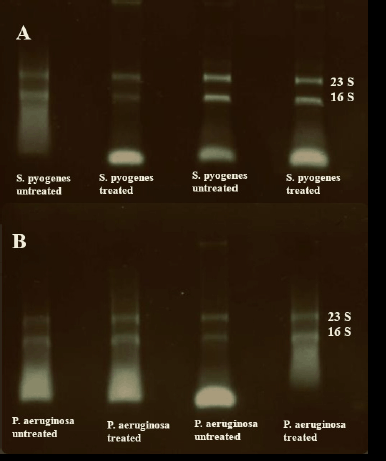 | Figure 3. Agarose gel electrophoresis of RNA samples. (A) Quality control for the RNA extracted from S. pyogenes biofilm. (B) Quality control for the RNA extracted from P. aeruginosa biofilm. [Click here to view] |
The findings of both RNA analysis methods, NanoDrop and the 2100 Bioanalyzer, were highly correlated, and the values were relevantly close. However, the readings of NanoDrop were relatively higher than the 2100 Bioanalyzer in a proportion of 1.1–2.5-fold, respectively. These results contradicted the results of the RT-PCR as when we unified the RNA template based on the readings of NanoDrop and the 2100 Bioanalyzer, there was a difference in the ct value of the amplified cDNA (Godoy et al., 2020). This highlights another issue of proper quantification of RNA content, specifically, that NanoDrop measures the double-stranded DNA and proteins in addition to RNA content. This is concomitant with the results of Hussing et al. (2018) reporting RNA concentrations are relevantly different according to the analysis method. In compliance with our results, several research groups reported a high-quality RNA extracted from various biological samples; for instance, RIN values between 8.75 and 9.9 were reported from bacterial cells (Heera et al., 2015), RIN value of 10 from yeast cells (Rodríguez and Vaneechoutte, 2019) (Rodríguez et al., 2020), and lower RIN values of 8 and 7 from human parotid tissue (Watermann et al., 2016) when using different kits for RNA extraction.
Moreover, recently, new methods emerged to examine RNA integrity such as DIV200 (Matsubara et al., 2020) and smear analysis methods (Anna Krowczynska, 2019). The DIV200 method is applied to examine the integrity of RNA molecules, whereas the RNA smear analysis determines if RNA molecules have a nucleotide size > 200 (Matsubara et al., 2020). There are several advantages of using DIV200 and smear analysis in testing RNA integrity over the RIN or gel electrophoresis, among them the ability to use both the DIV200 and smear analysis methods in testing the integrity of small yields of RNA extracted from biological tissue, whereas RIN integrity is useful in examining high RNA yields (Anna Krowczynska, 2019; Matsubara et al., 2020). We did not include the DIV200 and smear analysis methods in the RNA integrity examination as we achieved a high RNA yield in our experiments, and the RNA showed an intact texture when examined on gel electrophoresis.
 | Table 2. RNA purity (NanoDrop) and RNA integrity (2100 bioanalyzer) for different RNA extraction kits. [Click here to view] |
 | Figure 4. RIN for P. aeruginosa RNA extracted from untreated biofilms and biofilms treated with Trigona honey. (A) and (B) RIN for RNA extracted from untreated biofilms. C and D RIN for RNA extracted from treated biofilms with Trigona honey. [Click here to view] |
 | Figure 5. RNA RIN for S. pyogenes RNA extracted from untreated biofilms. (A) and (B) RIN for RNA extracted from untreated biofilms. C and D RIN for RNA extracted from treated biofilms with Trigona honey. [Click here to view] |
 | Table 3: RNA concentration (NanoDrop) and ct (RT PCR) for different RNA extraction kits. [Click here to view] |
CONCLUSION
We conclude that the usage of a single procedure in RNA extraction did not yield the required quality or quantity of RNA product. Therefore, the implementation of a mixture of solid-phase extraction and liquid-liquid extraction methods has augmented the high throughput of the RNA end product. Additionally, starting with a correct cellular number through performing a viable bacterial count is considered a limiting criterion in gene expression studies through conducting RT-PCR, microarray, or RNA sequencing.
CONFLICTS OF INTEREST
The author declares there are no conflicts of interest.
FUNDING
The author has conducted this research from private sources.
AUTHORS CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Atshan SS, Shamsudin MN, Lung LTT, Ling KH, Sekawi Z, Pei CP Ghaznavi-Rad E. Improved method for the isolation of RNA from bacteria refractory to disruption, including S. aureus producing biofilm. Gene, 2012; 494(2):219–24.
Bayatti N, Cooper-Knock J, Bury JJ, Wyles M, Heath PR, Kirby J Shaw PJ. Comparison of blood RNA extraction methods used for gene expression profiling in amyotrophic lateral sclerosis. PLoS One, 2014; 9(1):e87508.
Beltrame CO, Côrtes MF, Bandeira PT, Figueiredo AMS. Optimization of the RNeasy mini Kit to obtain high-quality total RNA from sessile cells of Staphylococcus aureus. Braz J Med Biol Res, 2015; 48:1071–6.
Brunet-Vega A, Pericay C, Quílez ME, Ramírez-Lázaro MJ, Calvet X, Lario S. Variability in microRNA recovery from plasma: comparison of five commercial kits. Anal Biochem, 2015; 488:28–35.
Buckingham L. Molecular diagnostics: fundamentals, methods and clinical applications. 3rd edition, FA Davis, Philadelphia, PA, 2019.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem, 2009; 55(4):611–22.
Cerca N, Pier GB, Vilanova M, Oliveira R, Azeredo J. Quantitative analysis of adhesion and biofilm formation on hydrophilic and hydrophobic surfaces of clinical isolates of Staphylococcus epidermidis. Res Microbiol, 2005; 156(4):506–14.
Cury JA, Koo H. Extraction and purification of total RNA from Sreptococcus mutans biofilms. Anal Biochem, 2007; 365(2):208–14.
Dolgin E. The tangled history of mRNA vaccines. Nature, 2021; 597(7876):318–24.
Godoy VCSMd, Bellucco FT, Colovati M, Oliveira-Junior HRd, Moysés-Oliveira M, Melaragno MI. Copy number variation (CNV) identification, interpretation, and database from Brazilian patients. Genet Mol Biol, 2020; 43:e20190218.
Grobe S, Doberenz S, Ferreira K, Krueger J, Bronstrup M, Kaever V, Haussler S. Identification and quantification of (t)RNA modifications in Pseudomonas aeruginosa by liquid chromatography-tandem mass spectrometry. Chembiochem, 2019; 20(11):1430–7.
Guttman K, Wang P, Jackson L, Morris A, Yau Y, Waters V. Isolation of total RNA from Pseudomonas aeruginosa within biofilms for measuring gene expression. J Vis Exp, 2021; doi:10.3791/62755(175).
Hassan A, Usman J, Kaleem F, Omair M, Khalid A, Iqbal M. Evaluation of different detection methods of biofilm formation in the clinical isolates. Braz J Infect Dis, 2011; 15(4):305–11.
He H, Li R, Chen Y, Pan P, Tong W, Dong X, Chen Y, Yu D. Integrated DNA and RNA extraction using magnetic beads from viral pathogens causing acute respiratory infections. Scientific reports, 2017; 7(1):1–8.
Heera R, Sivachandran P, Chinni SV, Mason J, Croft L, Ravichandran M, Yin LS. Efficient extraction of small and large RNAs in bacteria for excellent total RNA sequencing and comprehensive transcriptome analysis. BMC Res Notes, 2015; 8(1):1–11.
Hussing C, Kampmann M-L, Mogensen HS, Børsting C, Morling N. Quantification of massively parallel sequencing libraries–a comparative study of eight methods. Sci Rep, 2018; 8(1):1–9.
Imbeaud S, Graudens E, Boulanger V, Barlet X, Zaborski P, Eveno E, Mueller O, Schroeder A, Auffray C. Towards standardization of RNA quality assessment using user-independent classifiers of microcapillary electrophoresis traces. Nucleic Acids Res, 2005; 33(6):e56; doi: 10.1093/nar/gni054. PMID: 15800207; PMCID: PMC1072807.
Kafantaris I, Tsadila C, Nikolaidis M, Tsavea E, Dimitriou TG, Iliopoulos I, Amoutzias GD, Mossialos D. Transcriptomic analysis of Pseudomonas aeruginosa response to pine honey via RNA sequencing indicates multiple mechanisms of antibacterial activity. Foods, 2021; 10(5):936.
Kashofer K, Viertler C, Pichler M, Zatloukal K. Quality control of RNA preservation and extraction from paraffin-embedded tissue: implications for RT-PCR and microarray analysis. PLoS One, 2013; 8(7):e70714.
Kim ES, Lee JY, Park C, Ahn SJ, Bae HW, Cho YH. cDNA-derived RNA phage assembly reveals critical residues in the maturation protein of the Pseudomonas aeruginosa leviphage PP7. J Virol, 2021; 95(3):e01643–20.
Krowczynska A. Analysis of DNA fragments using the agilent 2100 bioanalyzer. Covaris, Inc.,Woburn, MA, 2019.
Le Rhun A, Beer YY, Reimegard J, Chylinski K, Charpentier E. RNA sequencing uncovers antisense RNAs and novel small RNAs in streptococcus pyogenes. RNA Biol, 2016; 13(2):177–95.
Liu X, Li Q, Wang X, Zhou X, Liao Q, He X, Zhang J, Sun J, Wu J, Cheng L. Comparison of six different pretreatment methods for blood RNA extraction. Biopreserv Biobank, 2015; 13(1):56–60.
Mack E, Neubauer A, Brendel C. Comparison of RNA yield from small cell populations sorted by flow cytometry applying different isolation procedures. Cytometry A, 2007; 71(6):404–9.
Maleševi? M, Di Lorenzo F, Filipi? B, Stanisavljevi? N, Novovi? K, Senerovic L, Polovi? N, Molinaro A, Koji? M, Jov?i? B. Pseudomonas aeruginosa quorum sensing inhibition by clinical isolate Delftia tsuruhatensis 11304: involvement of N-octadecanoylhomoserine lactones. Sci Rep, 2019; 9(1):1–13.
Manchester KL. Use of UV methods for measurement of protein and nucleic acid concentrations. Biotechniques, 1996; 20(6):968–70; doi: 10.2144/96206bm05. PMID: 8780864
Matsubara T, Soh J, Morita M, Uwabo T, Tomida S, Fujiwara T, Kanazawa S, Toyooka S, Hirasawa A. DV200 index for assessing RNA integrity in next-generation sequencing. Biomed Res Int, 2020; 2020:9349132.
Metcalf D, Weese JS. Evaluation of commercial kits for extraction of DNA and RNA from Clostridium difficile. Anaerobe, 2012; 18(6):608–13.
Mirani ZA, Fatima A, Urooj S, Aziz M, Khan MN, Abbas T. Relationship of cell surface hydrophobicity with biofilm formation and growth rate: a study on Pseudomonas aeruginosa, Staphylococcus aureus, and Escherichia coli. Iran J Basic Med Sci, 2018; 21(7):760.
Mommaerts K, Sanchez I, Betsou F, Mathieson W. Replacing β-mercaptoethanol in RNA extractions. Anal Biochem, 2015; 479:51–3.
Ngassam Tchamba C, Rao AS, Boyen F, Haesebrouck F, Duprez JN, Théron L, Thiry D, Mainil J. Comparison of quantitative PCR and MALDI-TOF mass spectrometry assays for identification of bacteria in milk samples from cows with subclinical mastitis. J Appl Microbiol, 2019; 127(3):683–92.
Peñuelas-Urquides K, Villarreal-Treviño L, Silva-Ramírez B, Rivadeneyra-Espinoza L, Said-Fernández S, León MBd. Measuring of Mycobacterium tuberculosis growth: a correlation of the optical measurements with colony forming units. Braz J Microbiol , 2013; 44(1):287–90.
Pionzio AM, McCord BR. The effect of internal control sequence and length on the response to PCR inhibition in real-time PCR quantitation. Forensic Sci Int Genet, 2014; 9:55–60.
Pusic P, Sonnleitner E, Blasi U. Specific and global RNA regulators in Pseudomonas aeruginosa. Int J Mol Sci, 2021; 22(16):8632.
Rayyan WA, Singh A, Al-Jaafreh A, Dayyih WA, Bustami M, Salem S, Seder N, Schröppel K. The role of glutamine-rich region of candida albicans Tec1p in mediating morphological transition and invasive growth. Int J Med Health Sci, 2019; 13(4):150–8.
Rodríguez A, Duyvejonck H, Van Belleghem JD, Gryp T, Van Simaey L, Vermeulen S, Van Mechelen E, Vaneechoutte M. Comparison of procedures for RNA-extraction from peripheral blood mononuclear cells. PLoS One, 2020; 15(2):e0229423.
Rodríguez A, Vaneechoutte M. Comparison of the efficiency of different cell lysis methods and different commercial methods for RNA extraction from Candida albicans stored in RNAlater. BMC Microbiol, 2019; 19(1):1–10.
Romero M, Silistre H, Lovelock L, Wright VJ, Chan KG, Hong KW, Williams P, Camara M, Heeb S. Genome-wide mapping of the RNA targets of the Pseudomonas aeruginosa riboregulatory protein RsmN. Nucleic Acids Res, 2018; 46(13):6823–40.
Sambrook J, Fritsch EF, Maniatis T. (1989). Molecular cloning: a laboratory manual (No. Ed. 2). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Sealfon SC, Chu TT. RNA and DNA microarrays. Methods Mol Biol, 2011; 671: 3–34.
Seder N, Abu Bakar MH, Abu Rayyan WS. Transcriptome analysis of Pseudomonas aeruginosa biofilm following the exposure to malaysian stingless bee honey. Adv Appl Bioinform Chem, 2021; 14:1–11.
Sonnleitner E, Prindl K, Blasi U. The Pseudomonas aeruginosa CrcZ RNA interferes with Hfq-mediated riboregulation. PLoS One, 2017; 12(7):e0180887.
Tavares L, Alves PM, Ferreira RB, Santos CN. Comparison of different methods for DNA-free RNA isolation from SK-N-MC neuroblastoma. BMC Res Notes, 2011; 4(1):1–5.
Thi MTT, Wibowo D, Rehm BH. Pseudomonas aeruginosa biofilms. Int J Mol Sci, 2020; 21(22):8671.
Watermann C, Peter Valerius K, Wagner S, Wittekindt C, Peter Klussmann J, Baumgart-Vogt E, Karnati S. Step-by-step protocol to perfuse and dissect the mouse parotid gland and isolation of high-quality RNA from murine and human parotid tissue. Biotechniques, 2016; 60(4):200–3.
Wei Q, Ma LZ. Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int J Mol Sci, 2013; 14(10):20983–1005.
Wong RK, MacMahon M, Woodside JV, Simpson DA. A comparison of RNA extraction and sequencing protocols for detection of small RNAs in plasma. BMC Genomics, 2019; 20(1):1–12.
Yip L, Fuhlbrigge R, Atkinson MA, Fathman CG. Impact of blood collection and processing on peripheral blood gene expression profiling in type 1 diabetes. BMC Genomics, 2017; 18(1):1–16.
Zhong Q, Yang L, Li L, Shen W, Li Y, Xu H, Zhong Z, Chen M, Le S. Transcriptomic analysis reveals the dependency of Pseudomonas aeruginosa genes for double-stranded RNA bacteriophage phiYY infection cycle. iScience, 2020; 23(9):101437.