
INTRODUCTION
Melanoma is the deadliest skin cancer that arises due to genetic mutations in cells that produce melanin, called melanocytes (Domingues et al., 2018). Melanoma represents less than 5% of all malignancies. However, most skin cancer deaths are caused by melanoma (Matthews et al., 2017). The mitogen-activated protein kinase (MAPK) pathway plays a role in cell proliferation-related cancer growth. Among the three isomers of Raf kinase (A-Raf, B-Raf, and C-Raf) in the MAPK pathway, B-Raf kinase is rated as the most crucial activator based on its mutation frequency (Mercer and Pritchard, 2003). Particularly in skin melanoma, the B-Raf kinase mutation rate ranges from 50% to 70%, and the most common is the conversion of valine to glutamic acid, known as B-RAF V600E (Luebker and Koepsell, 2019; Tuveson et al., 2003). In addition, several studies have also shown that mutations of valine to lysine (V600K) are also common (Jakob et al., 2012; Long et al., 2011). Therefore, B-Raf could be a potential target in developing anticancer drugs.
B-Raf inhibitors can be an option to treat metastatic melanoma (Belter et al., 2017). Two B-Raf inhibitor drugs that the FDA has approved for the treatment of B-RAF V600E/K mutant melanoma are vemurafenib and dabrafenib (Luebker and Koepsell, 2019). Administration of vemurafenib to patients with B-Raf V600 mutant metastatic melanoma showed excellent clinical response, and the mean overall survival was about 16 months (Sosman et al., 2012). Although the use of B-Raf inhibitors in the treatment of cancer has shown clinical success, there are side effects that lead to drug resistance, so it is necessary to find potential new entities, such as a B-Raf inhibitor (Zhang et al., 2017). The use of natural products as potential drugs could be an alternative option. More than 70% of medicinal products for clinical use come from natural products, and this also extends to cancerous cancers, which use natural products for chemotherapy drugs nowadays (Carranza and Newman, 2012).
The computational-based study is promising to be the first step in melanoma drug discovery. Technological developments in the field of molecular biology have discovered methods to identify protein structures. The discovery of these proteins supports computational methodologies to become a critical component in drug discovery and development strategies, from knowing hits to lead optimization and approaches such as ligand-based or virtual screening techniques (Meng et al., 2012; Wu et al., 2017). In general, the manufacture of drugs costs more than 1 billion USD and takes more than 10 years (Hughes et al., 2011). The development of computational or in silico methods reduces the number of molecules made and tested in vitro and in vivo; this kind of method helps relieve the time and costs needed (Ekins et al., 2007).
Microalgae have been known to produce various secondary and primary metabolites that could be developed as anticancer agents. They are considered sustainable aquatic resources that contain various active compounds (De Morais et al., 2015; Gastineau et al., 2014; Prasetiya et al., 2020, 2021). According to preliminary studies, several active compounds isolated from both freshwater and marine algae are known to repress the growth rate of cancer cells by increasing immunity and forming immunity to fight cancer cells (Fu et al., 2017; Gastineau et al., 2014; Jin et al., 2017; Martínez Andrade et al., 2018). As one of the biggest archipelago countries, Indonesia has numerous algae from various species. Unfortunately, they have not been used optimally (Hutomo and Moosa, 2005). Therefore, innovation is needed to optimize the use of microalgal resources, especially as therapeutic agents.
Previous in silico studies regarding inhibitor candidates for antimelanoma also used the B-Raf V600E as their target. However, these mostly focused on the design and synthesis of drug-like compounds and are limited to molecular docking simulations (Dong et al., 2013; Luo et al., 2008; Umar et al., 2021; Wang et al., 2017). Recent molecular dynamics studies showed the interactions of B-Raf V600E with synthetically designed compounds with less than 50 ns length simulations (Kaboli et al., 2018; Wu et al., 2021), while this research performed 200 ns length of simulations to observe their representative characteristics and interactions. Our study aimed to obtain insight into the B-Raf kinase receptor’s structure and identified which bioactive compounds can be used as potential inhibitors of B-Raf from microalgae. In addition, we retrieved a list of bioactive compounds from an established database and got a lead compound for the development of melanoma cancer drugs.
MATERIALS AND METHODS
Software and data collection
Molecular docking simulations were performed using the AutoDock 4.2 software downloaded from the Scripps Research Institute website (http://autodock.scripps.edu/). Visualization of the docking results was carried out with the BIOVIA Discovery Studio Visualizer 2021 and LigPlot+ (Laskowski and Swindells, 2011). The crystal structure of the B-Raf protein and its natural ligand (PDB ID: 4MNF,6Q0K) was retrieved from the Protein Data Bank (Haling et al., 2014; Park et al., 2019). The other 3D structures of ligands, the candidates of inhibitors, were obtained from PubChem. As for the drawing of the 2D structure, the ChemSketch 2021.1.2 (ACD/Labs) program was also used. Data collection strategies and the structure of compounds from microalgae potential as anticancer agents were carried out by searching the MarinLit database (http://pubs.rsc.org/marinlit/) and searching publications related to active compounds from aquatic organisms from 1970 to 2020 (Fig.1)
Homology modeling of B-Raf V600E
The alignment of the B-Raf V600E mutant protein sequence compared to the other B-Raf protein sequence from the Protein Data Bank (PDB ID: 4MNF, 6Q0K) has shown a gap in the sequence around the mutation. Therefore, this study modeled the full length of the native B-Raf receptor and the V600E mutant using Modeller 10.1 to investigate further anticancer melanoma candidates through molecular docking, virtual screening, and molecular dynamics simulation. PROCHECK (Laskowski et al., 1993) and the DOPE profile comparison of the model and template (Webb and Sali, 2016) were used to evaluate the B-Raf V600E models (Destiarani et al., 2020). These methods were able to rule out the protein structure with imprecise stereochemical features such as steric hindrance, improper hydrogen bonds, and distorted bond angles.
Ligand preparation and target receptor for molecular docking
The B-Raf V600E receptors and all ligands were stored in PDB format, including the control ligands, which are vemurafenib as a reference drug and GDC0879 as a natural ligand. The ligand molecule files were set aside, while the protein files were loaded into the AutoDockTools 1.5.6 software for further preparation steps. Protein preparation steps include the addition of polar hydrogen and Kollman charge, removing nonpolar hydrogen atoms, and converting to pdbqt format (Afriza et al., 2018). File conversion to pdbqt format was done to be used for molecular docking processes involving AutoGrid and AutoDock (Tao et al., 2020).
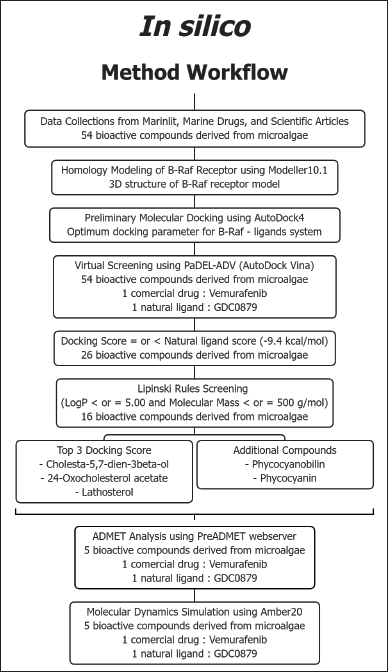 | Figure 1. The in silico method workflow for bioactive compounds screening candidates. [Click here to view] |
Preparation of the grid and molecular docking parameters
The parameter file grid was prepared with the help of AutoDockTools. To adjust the grid and center dimensions, amino acid residues located in the range of 5 Å around the residues which play an important role in substrate binding are selected. The grid box size was set to 20 × 20 × 20, coordinates were set to 18.944, −76.64, and 90.261 (as x, y, and z centers of mass, respectively), and spacing was set to 1.000 Å. Molecular anchoring was carried out using the Lamarck genetic algorithm (GA). A GA was used as the search parameter with the number of GA runs of 100, a population size of 150, and the maximum number of evaluations of 5,000,000 defined in the calculation. Grouping histograms, lowest binding energies, and root mean square deviation (RMSD) were then analyzed.
Preliminary molecular docking and virtual screening
Redocking and optimization were done using the AutoDock program to find the most appropriate coordinates and parameters on the active side. The parameters of the redocking results observed were amino acid residues and hydrogen bonds, and the RSMD value should be less than 2 Å. The Python scripts required for the preparation of potential compounds and their virtual screening are contained in AutoDockTools/Utilities24 and run using PaDEL-ADV. The docking results were then sorted based on the binding free energy of these compounds and also based on the most histogram groups. Afterward, the compound with the lowest binding energy was selected for further analysis of the interaction between the ligand compound and its receptor.
Lipinski rules screening
The bioactive compounds that mostly violated Lipinski’s rules (in terms of pharmacological characteristics) were discarded. According to Lipinski’s rules, a viable candidate for use as an orally active medicine should have no more than one violation of the following criteria: less than 5 hydrogen bond donors, less than 10 hydrogen bond acceptors, molecular weight less than 500 g/mol, and LogP less than 5 (Yang et al., 2020). The calculation of LogP was performed using the SwissADME webserver (http://www.swissadme.ch/index.php) with the iLOGP algorithm (Daina et al., 2014, 2017). However, the two additional compounds (phycocyanin and phycocyanobilin) which are produced massively from the most cultured microalgae species both in Indonesia and worldwide, such as Spirulina platina, Spirulina platensis, Spirulina sp., were also used for further analysis.
ADMET analysis
The PreADMET web server (https://preadmet.qsarhub.com/) was used to analyze anticancer melanoma candidates. Several parameters used in this ADMET assessment were HIA and Caco2 scores as absorption aspects, PPB and BBB as distribution aspects, inhibition ability of CYP2C19, CYP2C9, CYP2D6, and CYP3A4 as aspects of metabolism and excretion, and carcinogenic and mutagenic potential for aspects of toxicity.
Molecular dynamics simulation
Molecular dynamics simulations were carried out on the B-Raf protein system with inhibitor screening results on the B-RAF binding site. Molecular dynamics simulations were performed by the GPU with the program pmemd.cuda in Amber20 (Case et al., 2016). The notation of cysteine and histidine residues was adjusted according to their chemical environment. The TIP3P water molecule model was used as an explicit solvent with a minimum distance between the protein and the outer shell of 10 Å. The system was neutralized and set at a physiological salt concentration. Initial minimization was performed using 1,000 steps of steepest descent. Then, 2,000 steps of conjugate gradient minimization were carried out by applying 5 kcal/molÅ2 harmonic force resistance. The force resistance was gradually released from all heavy atoms (including side chains) to only atoms in the mainframe with the same minimization protocol. Finally, 5,000 steps of unimpeded conjugate gradient minimization were performed to eliminate spatial collisions. After that, the system was set to 310 K for mimicking the cellular environment in the human body, which is around 37°C (Khan et al., 2021; Rizo et al., 2022), in increments (0–100 K, 100–200 K, and 200–310 K for 20 ps each) for 60 ps. Next, the system density and pressure were equilibrated for 1,000 ps. The resistance force was released gradually during the equilibrium phase. Then, the production stage was carried out for 200 ns. The time step in the production phase was set to 2 fs. The MD simulation trajectories were then analyzed using the cpptraj module in AmberTools21.
Data analysis and visualization
Molecular dynamics simulation trajectories were analyzed visually with the help of VMD. RSMD, root mean square fluctuation (RMSF), and distance analysis were calculated using the cpptraj module in AmberTools21. The stability of the ligand in the B-Raf binding site was analyzed using the RMSD trajectory, while RMSF was used in the analysis of the stability of the B-Raf protein. The binding energy between the ligands in the B-Raf binding site was calculated using the program MMPBSA.py. Visualization of data was performed using the ggplot program in JupyterLab.
RESULTS AND DISCUSSION
Database of anticancer melanoma candidates from microalgae
A database of active compounds for anticancer melanoma candidates was constructed through literature studies from various databases such as MarinLit and Marine Drugs. In addition, data on active compounds were also obtained from international scientific articles on natural materials from marine waters (marine natural products) published from January 1970 to December 2019. The collected data were further classified based on the type of molecule, physical and chemical characteristics, microalgae source, and references. As supplementary information, the PubChem ID and the 3D structure of each molecule were also provided (Supplementary Table S1).
Virtual screening for inhibitors against B-Raf V600E candidates
The B-Raf V600E mutation in which the amino acid valine was substituted for glutamic acid is found in more than 60% of melanoma patients and can enhance MAPK signaling pathways independent of RAS activation (Davies et al., 2002; Wan et al., 2004). One of the first options for treating metastatic melanoma is B-Raf inhibitors (Belter et al., 2017). Purified B-Raf V600E is more active than fully activated B-Raf wild type, demonstrating that higher signaling output is required for transformation, according to steady-state kinetic studies. Despite the fact that the V600E mutation provides constitutive kinase activation independent of an intact side-to-side dimer interface, it has a larger possibility of forming oligomers. Furthermore, it avoids inhibitory p-loop phosphorylation in order to achieve the required increased signaling output for carcinogenesis (Cope et al., 2019). Hence, the objective of this study is to identify bioactive compounds from microalgae that are potential inhibitors against B-Raf V600E.
The preliminary molecular docking was carried out to collect prior data regarding the affinity between the B-Raf V600E receptor and its crystal structure ligand, GDC0879, and the generally recognized ligand as a melanoma cancer drug, vemurafenib. It has resulted in the lowest binding energy value of −8.33 kcal/mol and RMSD of 3.14 Å for the GDC0879 ligand and −8.95 kcal/mol and 3.95 Å for the vemurafenib ligand. Optimization steps are carried out to obtain parameter settings that produce the best binding energy values. This optimization process includes the variations in the volume of the grid box and the preparation of B-Raf receptors through minimization using the Amber20 program. The grid box is a critical factor for binding energy because it is the volume of the area involved in calculating and processing the energy value data itself (Feinstein and Brylinski, 2015). Additionally, the receptor preparation also aims to ease the structure of the B-Raf protein and minimize its energy, based on the literature that native structures in nature generally have the lowest free energies (Norn et al., 2021; Yu et al., 2020). The optimization of molecular docking with the vemurafenib ligand showed that the grid box 20 × 20 × 20:1 Å from the homology-modeled receptor resulted in the best binding energy value of −8.95 kcal/mol (Supplementary Table S2).
The virtual screening process was conducted using the PaDEL-ADV and AutoDock Vina programs on 54 active compounds derived from microalgae. The drug ligand vemurafenib was used as a standard and produced binding free energy of −10.4 kcal/mol so that only compounds with lower binding energy than this value could pass. In general, many of those compounds are the steroid group, while other compounds which also passed are phycocyanin and phycocyanobilin (Table 1). Based on a comparison of the docking scores between the candidate ligand for B-Raf V600E inhibitor and its natural ligand and the reference drug, the three best compounds are steroids, namely, cholesta-5,7-dien-3beta-ol, 24-oxocholesterol acetate, and lathosterol. The phycocyanin and phycocyanobilin were also analyzed because of their abundance as metabolites in Spirulina, one of Indonesia’s common cultured microalgae. In addition, some literature has also mentioned that phycocyanin has the potential as an anticancer drug candidate (Jiang et al., 2017; Liao et al., 2016; Ravi et al., 2015). If we follow Lipinski’s rule of five, both are not qualified candidates because their molecular weights exceed 500 g/mol. However, in general, several commercial drugs and potential drug candidates also do not pass this rule (Doak and Kihlberg, 2017; Ganesan, 2008). In fact, natural products are frequently mentioned as an exception to Lipinski’s rules, because nature has learned to preserve the hydrophobicity and intermolecular H-bond donor while producing biologically active molecules with high molecular weight and a large number of rotatable bonds (Ganesan, 2008; Newman and Cragg, 2020). For illustration, the two-dimensional interaction diagram from the docking of these ligands shows that the dominant interactions are hydrophobic (Table 2). Moreover, some of the amino acid residues on the binding site are also consistent with the predictions of the alanine scanning results (Supplementary Table S3). Another interaction involved is hydrogen bonds, where some atoms in the ligand can also act as hydrogen bond donors or acceptors.
ADME analysis and toxicity prediction
The analysis of ADME and its toxicity properties was carried out using the PreADMET program to reduce risks in drug development (Lee et al., 2003, 2004). The necessity for early assessment of ADME features is also becoming more pressing when combinatorial chemistry and high-throughput screening are implemented, which can create a large number of potential lead compounds (Boobis et al., 2002; Hodgson, 2001). Based on the absorption aspect, all candidates were well absorbed in the intestine, but on the Caco2 parameter, the three steroid compounds showed the highest permeability values (Table 3). In terms of distribution, PPB parameters showed that all candidates were soluble in blood and well distributed throughout the body, except for phycocyanin. However, naturally, phycocyanin binds to protein molecules (Grover et al., 2021; Padyana et al., 2001), so the possibility of being soluble in the blood is also high. Based on the dataset of 150 compounds, within this range, compounds with log BB > 0.40 cross the BBB readily while compounds with log BB < 0.40 are poorly distributed to the brain (Lobell et al., 2003; Ma et al., 2005). It is shown that the three steroid compounds have a high chance of crossing the brain barrier, and the same is for vemurafenib. Contrarily, phycocyanin, phycocyanobilin, and natural ligands will mostly not cross.
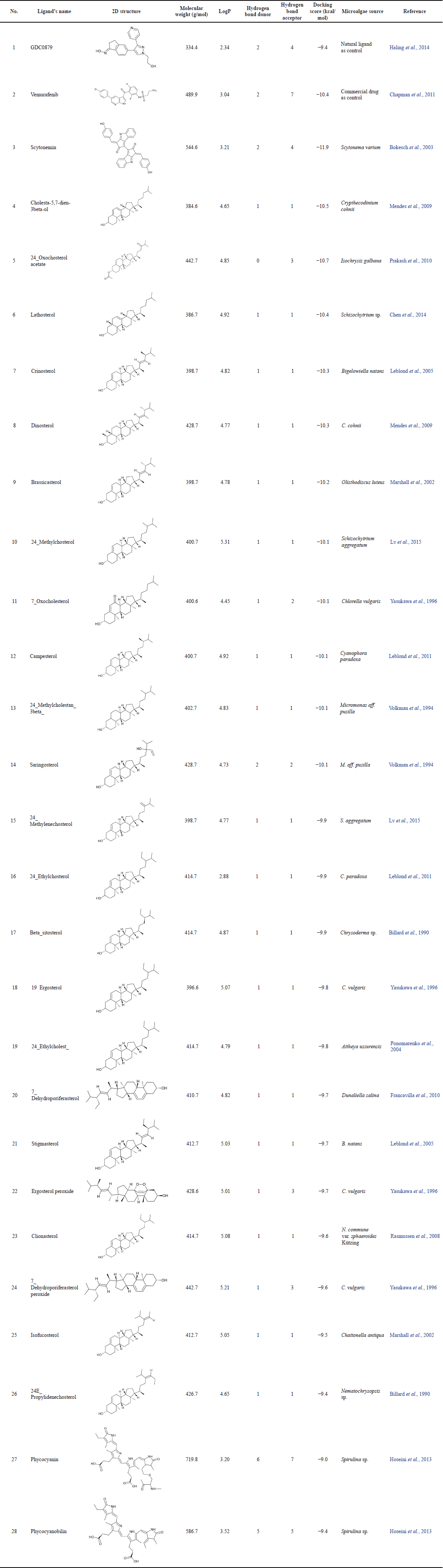 | Table 1. The results of virtual screening for melanoma anticancer compound candidates using the PaDEL-ADV program. [Click here to view] |
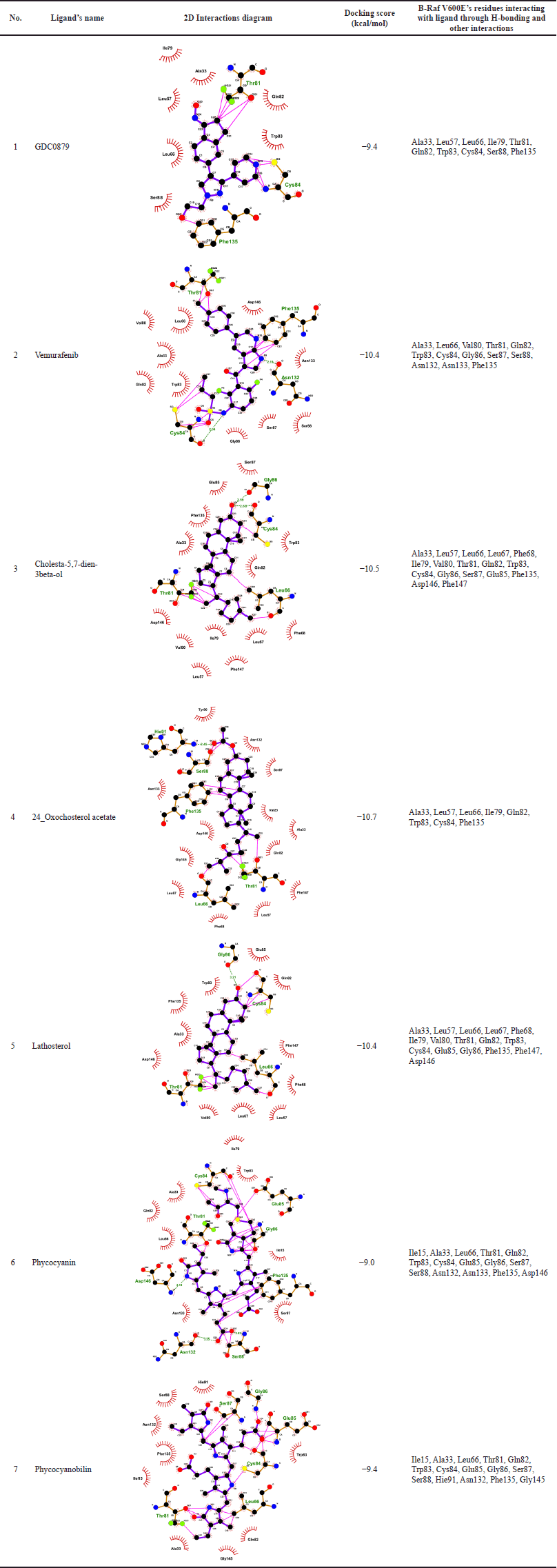 | Table 2. The interactions analysis of B-Raf V600E receptor and the binding of its ligands. [Click here to view] |
Analysis of candidate compounds as substrates for CYP or cytochrome P450, a class of enzymes involved in various drug metabolism processes, is needed to estimate possible metabolic processes (Lynch and Neff, 2007; McDonnell and Dang, 2013). As a substrate, candidate compounds could act as inducers that stimulate CYP and increase the activity of these enzymes. It can increase the metabolism of other substrates for CYP enzymes by reducing exposure to candidate compounds. Contrastingly, as an inhibitor that inhibits the activity of the CYP enzyme, it reduces the metabolism of other drugs that are substrates for the CYP enzyme to increase exposure to the candidate compound itself. Vemurafenib is a commercial drug that can act as a substrate for three of four types of CYP enzymes. The natural ligands and the three steroidal compounds can act as substrates for two types of enzymes, while phycocyanin and phycocyanobilin are only for one type. However, some drugs may not be metabolized by the CYP enzyme pathway (Dixit et al., 2017; Ghosal, 2020). The extensive use of metabolic stability screening in drug development, on the other hand, has resulted in the identification of new bioactive compounds that rely on non-CYP enzymes for clearance and also a rise in the number of pharmaceuticals that undergo metabolism via these enzymes (Gan et al., 2016). According to the most recent data, non-P450 enzymes play a substantial role in the 125 drugs’ metabolism. Around 30% of those drugs are metabolized by the specific non-P450 enzymes, which are aldehyde oxidase (1.1%), carbonyl reductases (2.4%), hydrolases (10.8%), and glucuronosyltransferases (11.7%) (Cerny, 2016).
Toxicity prediction is critical for public health. Among its numerous uses, it could significantly reduce the cost and labor of a medication’s preclinical and clinical trials, as many drug tests (cellular, animal, and clinical) can be avoided owing to projected toxicity (Wu and Wang, 2018). A toxicity test was carried out based on the mutagenicity predictions through the Ames test simulation and a 2-year simulation of the carcinogenicity test on mice and rats. The results showed that all candidate compounds except the natural ligand GDC0879 were non-mutagenic. Then, there are also positive results for vemurafenib, cholesta-5,7-dien-3beta-ol, and 24-Oxocholesterol acetate in terms of carcinogenicity. However, as genotoxicity test research progresses, it becomes clear that many of the drug candidates presently resulting in positive findings are either not carcinogenic or carcinogens with a nongenotoxic mechanism of action. This implies that many compounds have favorable genotoxicity information that is irrelevant to assess cancer risk. Some of them might be beneficial medications (Friedrich and Olejniczak, 2011; Walmsley and Billinton, 2011).
Molecular dynamics simulation assessment
RMSD analysis shows the average distance between atoms (usually the main chain) from the initial state, so it can be used to compare changes or shifts in protein conformation. Simulation results on the B-Raf V600E complex system with GDC0879 and vemurafenib showed a slight change in distance (Å) that is not too far from the initial conformation, which was approximately 2–3 Å. Stable conformation can be obtained if there is a strong enough interaction between the amino acid residues on the receptor and the functional groups on the ligands to keep the protein molecule compact so that the conformation is not much different from the initial state.
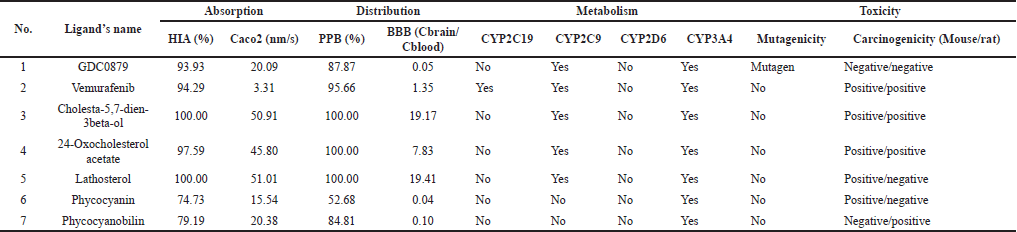 | Table 3. The results of ADME analysis and toxicity properties using the PreADMET program. [Click here to view] |
Generally, the system’s movement with ligands belonging to the steroid group (cholesta-5,7-dien-3beta-ol, 24-Oxocholesterol acetate, and lathosterol) is much higher than that of other systems, as indicated by an RMSD value of about 5–10 Å. Two other compounds, phycocyanin, and its derivative, phycocyanobilin, showed lower RMSD values, where the RMSD value pattern of phycocyanin was similar to the receptor system with its natural ligands. This shows that for 200 ns of dynamic simulation, a strong interaction occurs between the B-Raf V600E receptor and phycocyanin ligands (Fig. 2).
The calculation of RMSF is different from RMSD because it is carried out on each amino acid residue, each of which composes the structure of the native and mutant ND4L-ND6 protein subunits. The results of the RMSF analysis can provide information about the extent of the fluctuation of each residue during the simulation. This can describe the conformational shift of the amino acid residues that contributes to the flexibility of the protein. The RMSF value of steroid and vemurafenib ligands in certain residue areas is relatively higher than the B-Raf V600E receptor system with its natural ligand. On the other hand, both the phycocyanin and phycocyanobilin ligands showed lower RMSF values. Although in general, the RMSF value pattern of each ligand is similar to GDC0879, the natural ligand system, the drastic difference in RMSF values occurs in the residual region of the loop at the end of the chain so that the movement is much more accessible than in other regions (Fig. 3). The difference in the relatively higher RMSF value in the area at residues numbers 30–50 and 160–175 remained below the six range. This indicates that the overall dynamics of amino acid residues at the B-Raf V600E receptor tend to be stable. Based on the results of the alanine scanning analysis, several amino acid residues that are thought to have an important role are as follows: Lys35, Leu57, Leu66, Ile79, Trp83, Cys84, Phe135, and Phe147 (Supplementary Table S3). Some of these residues are also in the range of regions with relatively different RMSF values ??when compared to their natural ligand system.
The calculation of the binding free energy (ΔG°) between the B-Raf V600E receptor and each of its ligands was carried out using the molecular mechanics generalized born surface area (MMGBSA) method at every 20 ns interval during the molecular dynamics simulations. In addition, the calculation of the standard deviation and standard error was also included for each data collection. Estimating the correct binding mode and affinity of a ligand to its target protein can be done using the MMGBSA method. Molecular docking can introduce poses with the correct binding mode, and it also can be identified using other methods. However, a comparative analysis carried out on the binding free energy yields of molecular docking and MMGBSA showed that MMGBSA could improve the identification of different binding conformations by docking (Ahinko et al., 2019).
Based on the results of the binding free energy values?obtained, the phycocyanin ligand is considered to be the best candidate as an inhibitor because it has the lowest and most stable value, which is around −65 to −80 kcal/mol. As for natural ligands, commercial drugs and steroid-class ligands generally have almost the same value, which is around −40 to −60 kcal/mol, while phycocyanobilin has an even higher value of -40 to -30 kcal/mol (Fig. 4). The lower binding free energy value indicates that the binding that occurs is getting stronger and more favorable. Based on the analysis results, the interaction that generally plays a role in binding the ligand to the B-Raf V600E receptor is a hydrophobic interaction, where the binding site is also a hydrophobic region.
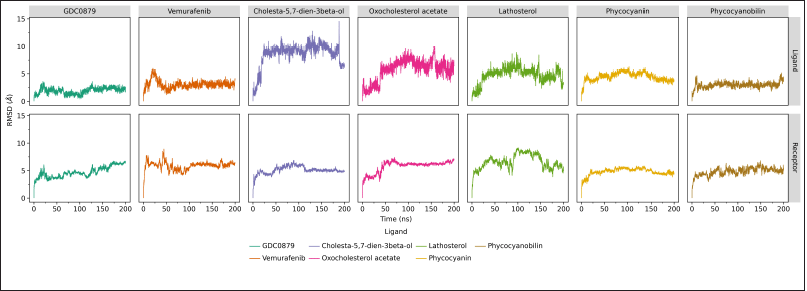 | Figure 2. RMSD analysis of 200 ns MD simulation of the B-Raf V600E receptor and each of its ligands, the natural ligands (GDC0879), commercial drugs (vemurafenib), and candidate inhibitor compounds calculated by cpptraj program in Amber20. Data visualization was created using the ggplot program on JupyterLab. [Click here to view] |
Distance analysis using the cpptraj program on AmberTools21 was carried out to show the average distance between the center of mass at the B-Raf V600E receptor and the center of mass of the ligands (Fig. 5). During the 200 ns molecular dynamics simulations, there will be a shift in the distance between the two, which is directly proportional to the stability of the binding. Our result demonstrated that the natural ligand GDC0879 and the phycocyanin ligand have a stable average distance of ~13 Å, where the density is almost close to one. Vemurafenib and the three steroid compounds tend to experience a distance shift throughout the simulation, while for phycocyanobilin, the distance is relatively stable but much further, which is around ~15 Å. The closer the average distance, the more stable the binding between the ligand and the receptor. Therefore, according to the binding free energy calculation results, phycocyanin also shows the closest distance.
The findings of this study have to be seen in the light of some limitations. For instance, the database size for bioactive compound candidates for antimelanoma is limited. This study presented the screening and prediction schemes to lead the examination of drug candidates. However, further in vitro analysis will be a proven concept and indeed complement these current results.
Previous molecular dynamics studies of B-Raf V600E revealed the destabilization of the B-Raf V600E inactive conformation but not in B-Raf wild type. The investigation of the interaction energies inside the catalytic region shows the presence of repulsive electrostatic forces acting on the activation loop of the mutant enzymes, commuting from inward to outward (Moretti et al., 2006). The V600E mutation disrupts the hydrophobic interactions in the wild-type structure, whereas the active state is maintained by forming a salt bridge between Glu600 and Lys507 (Maloney et al., 2021). The analysis of genes using the Comparative Toxicogenomics Database (CTD) depicted that B-Raf is one of 221 genes related to the specific melanoma disease and involved in activating the MAPK pathway and fibroblast growth factor receptor signaling pathway (Davis et al., 2021). Further, a more detailed analysis, such as the relations of the compounds to transcription factors and miRNAs involved, could be obtained using CTD, MicroRNA ENrichment TURned NETwork (http://userver.bio.uniroma1.it/apps/mienturnet), and the microRNA sponge generator and tester (http://www.med.muni.cz/histology/miRNAsong) (Duc Nguyen et al., 2022; Nguyen and Kim, 2022).
The RMSD pattern of phycocyanin against B-Raf V600E is consistent with the preceding simulations using aknadicine and 16beta-hydroxy-19svindolinine N-oxide as ligands; the most potent inhibitor will have the lowest RMSD value, which is the evidence of binding capability (Tang and Chen, 2015). Phycocyanin is indicated to have the activity to repress the growth of cancer cells as well as trigger an increase in immunity (Liu et al., 2016). Moreover, it is extracted from marine microalgae, which could be a good solution for the demand for sustainable products worldwide. Not to mention, Indonesia, as an archipelagic country, is rich in marine resources. As the major pigment compound in Spirulina, it is responsible for around 50% of light uptake in photosynthesis (Mishra et al., 2012; Queiroz et al., 2020) and has been shown to have reactive capacity against oxygen species, such as hydroxyl radical, alkoxyl radical, and peroxyl radical, that presents anti-inflammatory (Tripathi et al., 2021), neuroprotective, and hepatoprotective (Fernández-Rojas et al., 2014; Hamouda and El-Naggar, 2021) properties. Previous studies showed that phycocyanin was also valuable in managing diverse types of cancers. For example, it exhibited antitumor activity in carcinoma cells, triggering apoptosis by CD59 protein expression in HeLa cells (Yang et al., 2014). In addition, high-purity phycocyanin can induce the proliferation and differentiation of progenitor cells, also activating the macrophage function with the production of IL-I for the phagocytic process (Cherng et al., 2007). Thus, it becomes a favorable lead compound for drug development.
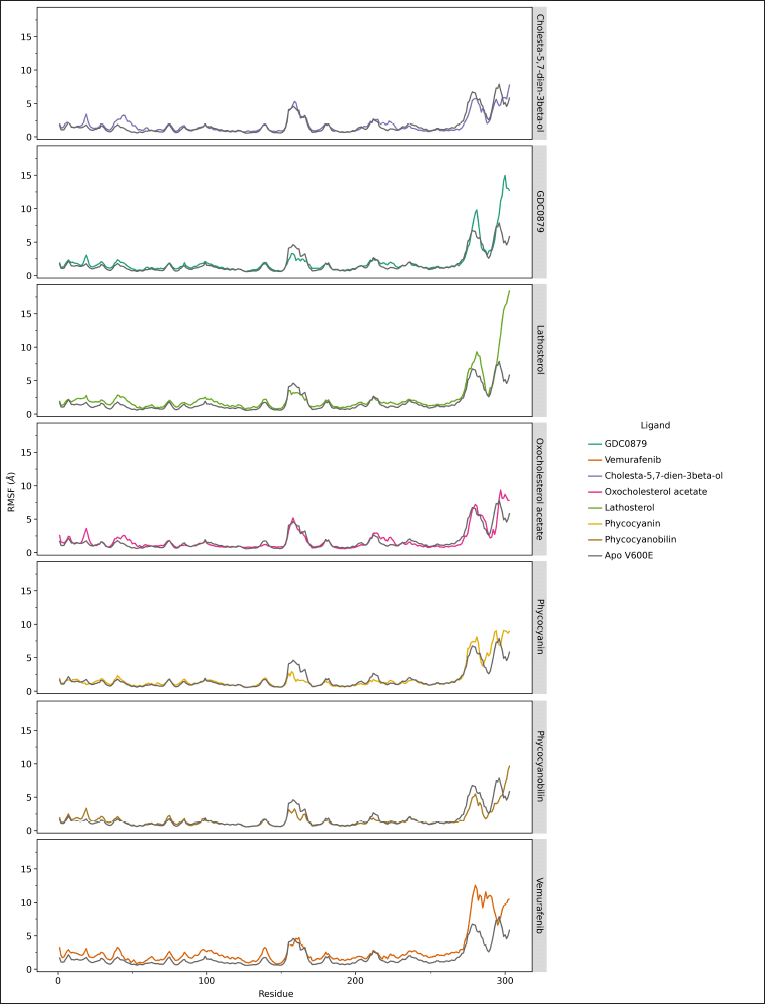 | Figure 3. RMSF analysis of the 303 amino acid residues making up the B-Raf V600E receptor during the 200 ns simulation. The difference between the native and mutant RMSF is indicated by the residue areas 30–50 and residues 160–175, as well as the end of the chain, namely, residues 280–303. Data visualization using the ggplot program in JupyterLab. [Click here to view] |
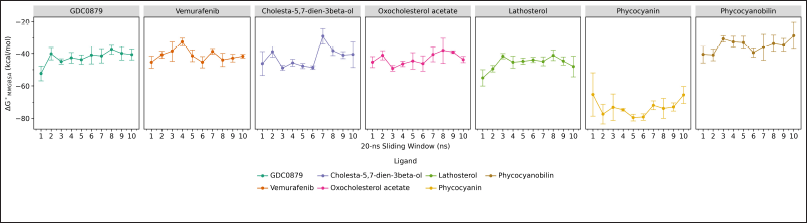 | Figure 4. The binding free energy of the B-Raf V600E receptor and each of its ligands, the natural ligands (GDC0879), commercial drugs (vemurafenib), and candidate inhibitor compounds calculated using MMGBSA method during 200 ns molecular dynamics simulations. Data visualization using the ggplot program in JupyterLab. [Click here to view] |
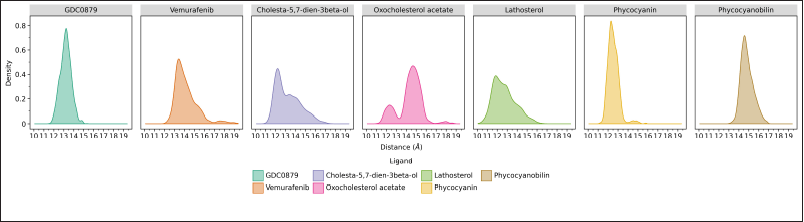 | Figure 5. Histogram of the distance between the B-Raf V600E receptor and its respective ligands for 200 ns of molecular dynamics simulation. The data was obtained based on calculations through the cpptraj program and visualized with the ggplot program on JupyterLab. [Click here to view] |
CONCLUSION
The candidates for active compounds produced by marine microalgae that have the potential to inhibit the growth of melanoma cancer cells have been collected in a new database. The prediction of the bioactivity of active compounds from marine algae as B-Raf receptor inhibitors on melanoma cancer cells using a virtual screening method resulted in the top three candidate compounds based on their docking score and the Lipinski rule, which are cholesta-5,7-dien-3beta-ol, 24-oxocholesterol acetate, and lathosterol. Phycocyanin and phycocyanobilin were also further examined because of their abundance in Spirulina sp., one of the most cultured microalgae in Indonesia. The molecular dynamics simulation showed that phycocyanin is the best candidate for melanoma anticancer drugs with free binding energy around −65 to −80 kcal/mol. It is also supported by the data of RMSD, RMSF, distance parameters, and many previous studies. This research provides a lead compound candidate that is expected to accelerate molecular research in developing therapeutic molecules for melanoma cancer.
ACKNOWLEDGMENTS
The authors would like to express their gratitude to the Research Center for Molecular Biotechnology and Bioinformatics, Universitas Padjadjaran, for their kind computational help and support. This work is supported by RPLK Research Grant No. 1959/UN6.3.1/PT.00/2021 from the Directorate of Research and Community Service (DRPM), Universitas Padjadjaran.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors declare there is no conflict of interest.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Afriza D, Suriyah WH, Ichwan SJA. In silico analysis of molecular interactions between the anti-apoptotic protein survivin and dentatin, nordentatin, and quercetin. J Phys Conf Ser, 2018; 1073(3):032001.
Ahinko M, Niinivehmas S, Jokinen E, Pentikäinen OT. Suitability of MMGBSA for the selection of correct ligand binding modes from docking results. Chem Biol Drug Des, 2019; 93(4):522–38.
Belter B, Haase-Kohn C, Pietzsch J. Biomarkers in malignant melanoma: recent trends and critical perspective. In: Ward WH, Farma JM (Eds.). Cutaneous melanoma: etiology and therapy, Codon Publications, Brisbane, AU, 2017.
Billard G, Dauguet JC, Maume D, Bert M. Sterols and chemotaxonomy of marine chrysophyceae. Bot Mar, 1990; 33(3):225–8.
Bokesch HR, O’Keefe BR, McKee TC, Pannell LK, Patterson GM, Gardella RS, Sowder RC 2nd, Turpin J, Watson K, Buckheit RW Jr, Boyd MR. A potent novel anti-HIV protein from the cultured cyanobacterium Scytonema varium. Biochemistry, 2003; 42(9):2578–84.
Boobis A, Gundert-Remy U, Kremers P, Macheras P, Pelkonen O. In silico prediction of ADME and pharmacokinetics. Eur J Pharm Sci, 2002; 17(4–5):183–93.
Case DA, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Xiao L, Kollman PA. Amber 2016 Reference Manual. University of California, San Francisco, CA, 2016.
Cerny MA. Prevalence of non-cytochrome P450-mediated metabolism in food and drug administration-approved oral and intravenous drugs: 2006–2015. Drug Metab Dispos, 2016; 44:1246–52.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New Engl J Med, 2011; 364(26):2507–16.
Chen J, Jiao R, Jiang Y, Bi Y, Chen ZY. Algal sterols are as effective as β-sitosterol in reducing plasma cholesterol concentration. J Agric Food Chem, 2014; 62(3):675–81.
Cherng SC, Cheng SN, Tarn A, Chou TC. Anti-inflammatory activity of c-phycocyanin in lipopolysaccharide-stimulated RAW 264.7 macrophages. Life Sci, 2007; 81(19–20):1431–5.
Cope N, Novak B, Candelora C, Wong K, Cavallo M, Gunderwala A, Liu Z, Li Y, Wang Z. Biochemical characterization of full-length oncogenic BRAFV600E together with molecular dynamics simulations provide insight into the activation and inhibition mechanisms of RAF kinases. Chem Bio Chem, 2019; 20(22):2850–61.
Daina A, Michielin O, Zoete V. ILOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model, 2014; 54(12):3284–301.
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7(1):1–13.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature, 2002; 417(6892):949–54.
Davis AP, Grondin CJ, Johnson RJ, Sciaky D, Wiegers J, Wiegers TC, Mattingly CJ. Comparative toxicogenomics database (CTD): update 2021. Nucleic Acids Res, 2021; 49:D1138–43.
De Morais MG, Vaz BDS, De Morais EG, Costa JAV. Biologically active metabolites synthesized by microalgae. BioMed Res Int, 2015; 2015:835761.
Destiarani W, Mulyani R, Yusuf M, Maksum IP. Molecular dynamics simulation of T10609C and C10676G mutations of mitochondrial ND4L gene associated with proton translocation in type 2 diabetes mellitus and cataract patients. Bioinform Biol Insights, 2020; 14:1177932220978672.
Dixit VA, Lal LA, Agrawal SR. Recent advances in the prediction of non-CYP450-mediated drug metabolism. Wiley Interdiscip Rev Comput Mol Sci, 2017; 7(6):e1323.
Doak BC, Kihlberg J. Drug discovery beyond the rule of 5—opportunities and challenges. Exp Opin Drug Discov, 2017; 12(2):115–9.
Domingues B, Lopes J, Soares P, Populo H. Melanoma treatment in review. Immunotargets Ther, 2018; 7:35–49.
Dong JJ, Li QS, Wang SF, Li CY, Zhao X, Qiu HY, Zhao MY, Zhu HL. Synthesis, biological evaluation and molecular docking of novel 5-phenyl-1H-pyrazol derivatives as potential BRAFV600E inhibitors. Org Biomol Chem, 2013; 11(37):6328–37.
Duc Nguyen H, Hee Jo W, Hong Minh Hoang N, Kim MS. Anti-inflammatory effects of B vitamins protect against tau hyperphosphorylation and cognitive impairment induced by 1,2 diacetyl benzene: an in vitro and in silico study. Int Immunopharmacol, 2022; 108:108736.
Carranza EA, Newman M. Clinical periodontology. 9th edition, W. B. Saunders, Philadelphia, PA, 2012.
Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol, 2007; 152(1):21–37.
Feinstein WP, Brylinski M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J Cheminform, 2015; 7(1):18.
Fernández-Rojas B, Hernández-Juárez J, Pedraza-Chaverri J. Nutraceutical properties of phycocyanin. J Funct Foods, 2014; 11:375–92.
Francavilla M, Trotta P, Luque R. Phytosterols from Dunaliella tertiolecta and Dunaliella salina: a potentially novel industrial application. Bioresour Technol, 2010; 101(11):4144–50.
Friedrich A, Olejniczak K. Evaluation of carcinogenicity studies of medicinal products for human use authorised via the European centralised procedure (1995-2009). Regul Toxicol Pharmacol, 2011; 60(2):225–48.
Fu W, Nelson DR, Yi Z, Xu M, Khraiwesh B, Jijakli K, Chaiboonchoe A, Alzahmi A, Al-Khair D, Brynjolfsson S, Salehi-Ashtiani K. Bioactive compounds from microalgae: current development and prospects. Studies in natural products chemistry. Elsevier, pp 199–225, 2017.
Gan J, Ma S, Zhang D. Non-cytochrome P450-mediated bioactivation and its toxicological relevance. Drug Metab Rev, 2016; 48(4):473–501.
Ganesan A. The impact of natural products upon modern drug discovery. Curr Opin Chem Biol, 2008; 12(3):306–17.
Gastineau R, Turcotte F, Pouvreau JB, Morançais M, Fleurence J, Windarto E, Prasetiya FS, Arsad S, Jaouen P, Babin M, Coiffard L, Couteau C, Bardeau JF, Jacquette B, Leignel V, Hardivillier Y, Marcotte I, Bourgougnon N, Tremblay R, Deschênes JS, Badawy H, Pasetto P, Davidovich N, Hansen G, Dittmer J, Mouget JL. Marennine, promising blue pigments from a widespread Haslea diatom species complex. Mar Drugs, 2014; 12(6):3161–89.
Ghosal A. Evaluation of the clearance mechanism of non-CYP-mediated drug metabolism and DDI as a victim drug. In: Ma S, Chowdhury S (Eds.). Identification and quantification of drugs, metabolites, drug metabolizing enzymes, and transporters. Elsevier Science, pp 237–71, 2020.
Grover P, Bhatnagar A, Kumari N, Narayan Bhatt A, Kumar Nishad D, Purkayastha J. C-Phycocyanin-a novel protein from Spirulina platensis—in vivo toxicity, antioxidant and immunomodulatory studies. Saudi J Biol Sci, 2021; 28(3):1853–9.
Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung Wç, Bravo BJ, Giannetti AM, Peck A, Masselot A, Morales T, Smith D, Brandhuber BJ, Hymowitz SG, Malek S. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell, 2014; 26(3):402–13.
Hamouda RA, El-Naggar NEA. Cyanobacteria-based microbial cell factories for production of industrial products. In: Singh V (Ed.). Microbial cell factories engineering for production of biomolecules, Elsevier, pp 277–302, 2021.
Hodgson J. ADMET—turning chemicals into drugs. Nature Biotechnol, 2001; 19(8):722–6.
Hoseini SM, Khosravi-Darani K, Mozafari MR. Nutritional and medical applications of Spirulina microalgae. Mini Rev Med Chem, 2013; 13(8):1231–7.
Hughes JP, Rees SS, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol, 2011; 162(6):1239–49.
Hutomo M, Moosa MK. Indonesian marine and coastal biodiversity: present status. Indian J Mar Sci, 2005; 34(1):88–97.
Jakob JA, Bassett RL Jr, Ng CS, Curry JL, Joseph RW, Alvarado GC, Rohlfs ML, Richard J, Gershenwald JE, Kim KB, Lazar AJ, Hwu P, Davies MA. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer, 2012; 118(16):4014–23.
Jiang L, Wang Y, Yin Q, Liu G, Liu H, Huang Y, Li B. Phycocyanin: a potential drug for cancer treatment. J Cancer, 2017; 8(17):3416–29.
Jin W, Liu G, Zhong W, Sun C, Zhang Q. Polysaccharides from Sargassum thunbergii?: monthly variations and anti-complement and anti-tumour. Int J Biol Macromol, 2017; 105:1526–31
Kaboli PJ, Ismail P, Ling KH. Molecular modeling, dynamics simulations, and binding efficiency of berberine derivatives: a new group of RAF inhibitors for cancer treatment. PLoS One, 2018; 13(3):1–19.
Khan T, Khan AR, Raza S, Azad I, Lawrence AJ. Medicinal and environmental chemistry: experimental advances and simulations. Bentham Science Publisher, 2021.
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr, 1993; 26(2):283–91.
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model, 2011; 51:2778–86.
Leblond JD, Dahmen JL, Seipelt RL, Elrod-Erickson MJ, Kincaid R, Howard JC, Evens TJ, Chapman PJ. Lipid composition of chlorarachniophytes (Chlorarachniophyceae) from the genera Bigelowiella, Gymnochlora, and Lotharella. J Phycol, 2005; 41(2):311–21.
Leblond JD, Timofte HI, Roche SA, Porter NM. Sterols of glaucocystophytes. Phycol Res, 2011; 59(2):129–34.
Lee S, Chang GS, Lee IH, Chung JE, Sung KY, No K. The PreADME: PC-based program for batch prediction of ADME properties. EuroQSAR 2004, 2004; 9:5–10.
Lee SK, Lee IH, Kim HJ, Chang GS, Chung JE, No KT. The PreADME approach: web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. EuroQSAR 2002 designing drugs and crop protectants: processes, problems and solutions. Blackwell Publishing, Malden, MA, pp 418–420, 2003.
Liao G, Gao B, Gao Y, Yang X, Cheng X, Ou Y. Phycocyanin inhibits tumorigenic potential of pancreatic cancer cells: role of apoptosis and autophagy. Sci Rep, 2016; 6:1–12.
Liu Q, Huang Y, Zhang R, Cai T, Cai Y. Medical application of Spirulina platensis derived C-phycocyanin. Evid Based Complement Alternat Med, 2016; 2016:7803846.
Lobell M, Molnár L, Keserü GM. Recent advances in the prediction of blood-brain partitioning from molecular structure. J Pharm Sci, 2003; 92(2):360–70.
Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol, 2011; 29(10):1239–46.
Luebker SA, Koepsell SA. Diverse mechanisms of BRAF inhibitor resistance in melanoma identified in clinical and preclinical studies. Front Oncol, 2019; 9:1–8.
Luo C, Xie P, Marmorstein R. Identification of BRAF inhibitors through in silico screening. J Med Chem, 2008; 51:6121–7.
Lv J, Yang X, Ma H, Hu X, Wei Y, Zhou W, Li L. The oxidative stability of microalgae oil (Schizochytrium aggregatum) and its antioxidant activity after simulated gastrointestinal digestion: relationship with constituents. Eur J Lipid Sci Technol, 2015; 117(12):1928–39.
Lynch T, Neff AP. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Phys, 2007; 76(3):391–6.
Ma XL, Chen C, Yang J. Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharmacol Sin, 2005; 26(4):500–12.
Maloney RC, Zhang M, Jang H, Nussinov R. The mechanism of activation of monomeric B-Raf V600E. Comput Struct Biotechnol J, 2021; 19:3349–63.
Marshall JA, Nichols PD, Hallegraeff GM. Chemotaxonomic survey of sterols and fatty acids in six marine raphidophyte algae. J Appl Phycol, 2002; 14(4):255–65.
Martínez Andrade KA, Lauritano C, Romano G, Ianora A. Marine microalgae with anti-cancer properties. Mar Drugs, 2018; 16(5):165.
Matthews NH, Li WQ, Qureshi AA, Weinstock MA, Cho E. Epidemiology of melanoma. Progress in clinical cancer. vol. 6, pp 139–49, 2017.
McDonnell AM, Dang CH. Basic review of the cytochrome P450 system. J Adv Pract Oncol, 2013; 4(4):263.
Mendes A, Reis A, Vasconcelos R, Guerra P, Lopes Da Silva T. Crypthecodinium cohnii with emphasis on DHA production: a review. J Appl Phycol, 2009; 21(2):199–214.
Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des, 2012; 7(2):146–57.
Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta Rev Cancer, 2003; 1653(1):25–40.
Mishra SK, Shrivastav A, Maurya RR, Patidar SK, Haldar S, Mishra S. Effect of light quality on the C-phycoerythrin production in marine cyanobacteria Pseudanabaena sp. isolated from Gujarat coast, India. Protein Expr Purif, 2012; 81:5–10.
Moretti S, Macchiarulo A, De Falco V, Avenia N, Barbi F, Carta C, Cavaliere A, Melillo RM, Passeri L, Santeusanio F, Tartaglia M, Santoro M, Puxeddu E. Biochemical and molecular characterization of the novel BRAF V599Ins mutation detected in a classic papillary thyroid carcinoma. Oncogene, 2006; 25(30):4235–40.
Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod, 2020; 83(3):770–803.
Nguyen HD, Kim MS. The protective effects of curcumin on metabolic syndrome and its components: in-silico analysis for genes, transcription factors, and microRNAs involved. Arch Biochem Biophys, 2022; 727:109326.
Norn C, Wicky BIM, Juergens D, Liu S, Kim D, Tischer D, Koepnick B, Anishchenko I; Foldit Players, Baker D, Ovchinnikov S. Protein sequence design by conformational landscape optimization. Proc Natl Acad Sci U S A, 2021; 118(11):1–7.
Padyana AK, Bhat VB, Madyastha KM, Rajashankar KR, Ramakumar S. Crystal structure of a light-harvesting protein C-phycocyanin from Spirulina platensis. Biochem Biophys Res Commun, 2001; 282(4):893–8.
Park E, Rawson S, Li K, Kim BW, Ficarro SB, Pino GG, Sharif H, Marto JA, Jeon H, Eck MJ. Architecture of autoinhibited and active BRAF–MEK1–14-3-3 complexes. Nature, 2019; 575(7783):545–50.
Ponomarenko LP, Stonik IV, Aizdaicher NA, Orlova TY, Popovskaya GI, Pomazkina GV, Stonik VA. Sterols of marine microalgae Pyramimonas cf. cordata (Prasinophyta), Attheya ussurensis sp. nov. (Bacillariophyta) and a spring diatom bloom from Lake Baikal. Comp Biochem Physiol B Biochem Mol Biol, 2004; 138(1):65–70.
Prakash S, Sasikala SL, Aldous VHJ. Isolation and identification of MDR-Mycobacterium tuberculosis and screening of partially characterised antimycobacterial compounds from chosen marine micro algae. Asian Pac J Trop Med, 2010; 3(8):655–61.
Prasetiya FS, Ramdhani DS, Mulyani Y, Mochamad U, Agung K, Arsad S, Mouget J. Antioxidant activities of culture supernatant Haslea ostrearia adapted in Indonesia. AACL Bioflux, 2021; 14(1):111–9.
Prasetiya FS, Sunarto S, Bachtiar E, Agung MUK, Nathanael B, Pambudi AC, Lestari AD, Astuty S, Mouget J. Effect of the blue pigment produced by the tropical diatom Haslea nusantara on marine organisms from different trophic levels and its bioactivity. Aquac Rep, 2020; 17:100389.
Queiroz MI, Vieira JG, Maroneze MM. Morphophysiological, structural, and metabolic aspects of microalgae. Elsevier Inc., 2020.
Rasmussen HE, Blobaum KR, Park YK, Ehlers SJ, Lu F, Lee JY. Lipid extract of Nostoc commune var. sphaeroides Kützing, a blue-green alga, inhibits the activation of sterol regulatory element binding proteins in HepG2 cells. J Nutr, 2008; 138(3):476–81.
Ravi M, Tentu S, Baskar G, Rohan Prasad S, Raghavan S, Jayaprakash P, Jeyakanthan J, Rayala SK, Venkatraman G. Molecular mechanism of anti-cancer activity of phycocyanin in triple-negative breast cancer cells. BMC Cancer, 2015; 15(1):1–13.
Rizo J, Sari L, Qi Y, Im W, Lin MM. All-atom molecular dynamics simulations of Synaptotagmin-SNARE-complexin complexes bridging a vesicle and a flat lipid bilayer. eLife, 2022; 11:1–28.
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Hersey P, Kefford R, Lawrence D, Puzanov I, Lewis KD, Amaravadi RK, Chmielowski B, Lawrence HJ, Shyr Y, Ye F, Li J, Nolop KB, Lee RJ, Joe AK, Ribas A. survival in BRAF V600—mutant advanced melanoma treated with vemurafenib. N Engl J Med, 2012; 366(8):707–14.
Tang HSC, Chen YC. Insight into molecular dynamics simulation of BRAF(V600E) and potent novel inhibitors for malignant melanoma. Int J Nanomed, 2015; 10:3131–46.
Tao X, Huang Y, Wang C, Chen F, Yang L, Ling L, Che Z, Chen X. Recent developments in molecular docking technology applied in food science: a review. Int J Food Sci Technol, 2020; 55(1):33–45.
Tripathi R, Shalini R, Singh RK. Prophyletic origin of algae as potential repository of anticancer compounds. Evolutionary diversity as a source for anticancer molecules. Academic Press, pp 155–89, 2021.
Tuveson DA, Weber BL, Herlyn M. BRAF as a potential therapeutic target in melanoma and other malignancies. Cancer Cell, 2003; 4(2):95–8.
Umar AB, Uzairu A, Shallangwa GA, Uba S. Molecular docking strategy to design novel V600E-BRAF kinase inhibitors with prediction of their drug-likeness and pharmacokinetics ADMET properties. Chem Afr, 2021; 4(1):189–205.
Volkman JK, Barrett SM, Dunstan GA, Jeffrey SW. Sterol biomarkers for microalgae from the green algal class Prasinophyceae. Org Geochem, 1994; 21(12):1211–8.
Walmsley RM, Billinton N. How accurate is in vitro prediction of carcinogenicity? Br J Pharmacol, 2011; 162(6):1250–8.
Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R, Cancer Genome Project. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell, 2004; 116(6):855–67.
Wang GM, Wang X, Zhu JM, Guo BB, Yang Z, Xu ZJ, Li B, Wang HY, Meng LH, Zhu WL, Ding J. Docking-based structural splicing and reassembly strategy to develop novel deazapurine derivatives as potent B-Raf V600E inhibitors. Acta Pharmacol Sin, 2017; 38:1059–68.
Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics, 2016; 2016:5.6.1–37.
Wu B, Zhang Z, Dou G, Lv X, Ge J, Wang H, Xie H, Zhu D. Novel natural inhibitors targeting B-RAF(V600E) by computational study. Bioengineered, 2021; 12(1):2970–83.
Wu CH, Arighi C, Ross K. Protein bioinformatics: from protein modifications and networks to proteomics. Methods in molecular biology. Humana Press, New York, NY, vol. 1558, p 472, 2017.
Wu Y, Wang G. Machine learning based toxicity prediction: from chemical structural description to transcriptome analysis. Int J Mol Sci, 2018; 19(8):2358.
Yang F, Li B, Chu XM, Lv CY, Xu YJ, Yang P. Molecular mechanism of inhibitory effects of C-phycocyanin combined with all-trans-retinoic acid on the growth of HeLa cells in vitro. Tumor Biol, 2014; 35(6):5619–28.
Yang Y, Shi CY, Xie J, Dai JH, He SL, Tian Y. Identification of potential dipeptidyl peptidase (DPP)-IV inhibitors among moringa oleifera phytochemicals by virtual screening, molecular docking analysis, ADME/T-based prediction, and in vitro analyses. Molecules, 2020; 25(1):189.
Yasukawa K, Akihisa T, Kanno H, Kaminaga T, Izumida M, Sakoh T, Tamura T, Takido M. Inhibitory effects of sterols isolated from Chlorella vulgaris on 12-0-tetradecanoylphorbol-13-acetate-induced inflammation and tumor promotion in mouse skin. Biol Pharm Bull, 1996; 19(4):573–6.
Yu M, Tan P, Ye Y, Voneshen DJ, Xing X, Hong L. One-dimensional nature of protein low-energy vibrations. Phys Rev Res, 2020; 2(3):1–6.
Zhang W, Qiu KX, Yu F, Xie XG, Zhang SQ, Chen YJ, Xie HD. Virtual screening of B-Raf kinase inhibitors: a combination of pharmacophore modelling, molecular docking, 3D-QSAR model and binding free energy calculation studies. Comput Biol Chem, 2017; 70:186–90.