
INTRODUCTION
Antiviral drug nevirapine is one of the preferred choices for the treatment of AIDS and is effectively used in Highly active antiretroviral therapy (HAART) for the management of the disease. It stops the growth of the virus at a submicromolar concentration by inhibiting the ability of the viral enzyme to transcribe the ribonucleic acid (Jindal and Mehta 2015; Wang et al., 2019). The major concern with nevirapine is its side effects which include Central nervous system (CNS) toxicity, hepatotoxicity, insomnia, depression, nausea, rashes, and hyperlipidemia. It gets metabolized by CYP3A and stimulates mutagenicity and hepatocarcinogenicity (Kuo and Chung, 2011). Also, a high dose has to be given to penetrate viral reservoir sites, which results in intolerance (Shegokar and Singh, 2011).
In addition to the mentioned toxicity, nevirapine belongs to the Biopharmaceutics Classification System (BCS) class II drugs for the reasons of poor water solubility (~0.1 mg/ml)and pH-dependent solubility with a pKa of 2.8 (Chadha et al., 2010; Moretton et al., 2014). In an acid medium, the solubility is 4.99 g/l and the solubility is reduced by a factor of 10–40 when pH is increased. This observable fact is due to change in the structure of molecules at different pH (de Oliveira et al., 2013). Solubility plays a significant role in in-vitro and in-vivo performances of a drug, and dissolution of a drug depends on ionic strength, pH, and buffer capacity of the Gastro intestine (GI) fluids (Hamed et al., 2016). Due to the above-mentioned reasons, the dissolution of nevirapine has been considered to be the rate-limiting step for its absorption.
Always there exists a correlation between solubility and bioavailability. A number of formulation strategies could be used to improve the solubility of class II drugs, such as a change in crystal habit, particle size reduction, chemical approaches, and formulation approaches using carriers (Leuner and Dressman, 2000; Vasconcelos et al., 2016). Solid dispersion (SD) is one of the approaches, which enhances solubility by reducing particle size and increasing wettability (Huang and Dai, 2014). Among the four generations of SD, the third generation SD is prepared using surfactants as carriers; this has the advantage of increased drug release, long stability, and enhanced bioavailability (Vo et al., 2013).
An advance in formulation technology has introduced a range of carriers for the preparation of dispersion. Gelucire belongs to the group of saturated polyglycolized glycerides that has gained more attention as SD carriers due to its surface active properties. It reduces the surface tension between the drug and dissolution medium and consequently enhances the oral bioavailability of the poorly soluble drug (Bertoni et al., 2020; da Fonseca Antunes et al., 2013). It has been successfully used as a carrier in SD of antiviral drugs (Damian et al., 2000; Sinha et al., 2010), ursolic acid (de Oliveira Eloy et al., 2012), carvedilol (Potluri et al., 2011), and Lycopene (Faisal et al., 2013).
In recent years, another approach has gained importance in the delivery of poorly soluble drug called lipid-based drug delivery system, which can enhance solubility and oral bioavailability (Gershanik and Benita, 2000; Sandeep et al., 2013; Singh et al., 2020). Self-emulsifying drug delivery system (SEDDS) is one among them, consisting of a blend of oils, surfactants, and co-surfactants, on mild agitation with GI fluid forms o/w microemulsion, and both hydrophilic and hydrophobic drugs can be incorporated (Gursoy and Benita, 2004; Jing-ling et al., 2007; Nardin and Köllner, 2019). In addition to solubility and bioavailability, it enhances drug uptake by the intestinal lymphatic system and protects from the hostile environment in the gut (Colin, 1997; Jing-ling et al., 2007; Krishnamoorthy et al., 2013; Sandeep et al., 2013).
In the present study, we have investigated a novel self-dispersing lipid formulation (SDLF) of nevirapine by combining the benefits of both SD and self-emulsifying system (SES). As SES has the advantage of overcoming toxicity associated with the drug, initially SES was designed using various combinations of oil, surfactant, and co-solvent and was characterized for drug loading, microscopic, and self-emulsifying properties. Later, a SD of drug was prepared using gelucire 50/13 as a carrier to improve its solubility and characterized for drug using DSC, XRD, and in-vitro drug release. Subsequently, novel SDLF was developed by combining SD and SES and evaluated for its in-vitro and in-vivo behavior.
MATERIALS AND METHODS
Nevirapine was a gift sample from Micro Labs Limited, Bengaluru, India. Gelucire 50/13 and Transcutol HP were gift samples from Gattefosee India Pvt. Ltd, Mumbai, India. Neucil US2 and Cremophor RH 40 were gift samples from Strides Arco Labs, Bengaluru, India. Oleic acid was purchased from SD Fine Chemicals, Mumbai, India. All the chemicals used in this study were analytical grade. Buffers were prepared using double distilled water.
Preparation and characterization of drug-loaded SES
Oil (oleic acid), surfactant (Cremophor RH 40), and co-surfactant (Transcutol HP) were selected based on drug solubility and emulsifying ability. Nevirapine-SES (NV-SES) formulations were prepared by dissolving nevirapine in different amounts of oil (10%–90%) placed in glass vials at ambient temperature by vortexing on a vortex mixer. To the mixture, previously prepared surfactant co-surfactant mixture (S/CoS mix) in the ratio of 3:1 was added in the range of 10%–90% (Table 1) and vortexed with simultaneous heating up to 40°C until a homogenous transparent preparation was obtained. NV-SES formulations were evaluated for drug loading, phase separation, precipitation, and the emulsifying ability for selecting optimized NV-SES (Cometa et al., 2020; Komal et al., 2015; Shweta et al., 2011). Optimized NV-SES was subjected to aqueous dilution to get microemulsion and evaluated for droplet size, polydispersity index (PDI), and zeta potential (ZP) using Malvern Zetasizer instrument (Nano-ZS, UK). The solubility of the drug in excipients and SES was determined using either DMSO/Methanol as diluent by UV spectrophotometer (Shimadzu, Pharmaspec UV-1700) at wavelength 282 nm.
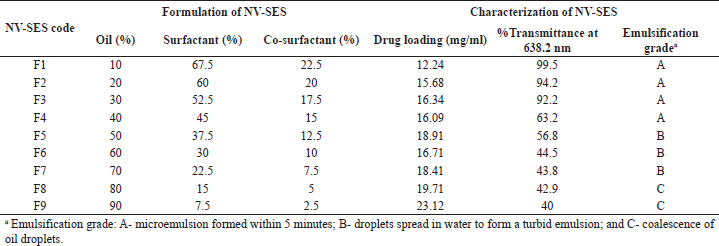 | Table 1. Formulation and characterization of NV-SES. [Click here to view] |
Preparation and characterization of drug-loaded SDs
A SD of nevirapine with Gelucire 50/13 was prepared in the ratio of 1:1, 1:2, 1:3, and 1:4 by the conventional melt method. Nevirapine was dissolved in a small quantity of ethanol and this solution was added to melt gelucire 50/13 (at 50°C) in a china dish. The solution mixture was dried in a rotary evaporator to remove ethanol and cooled rapidly in an ice bath (Faisal et al., 2013; Sinha et al., 2010). Prepared SDs were subjected to in-vitro drug release in intestinal pH 6.8. The selected ratio was subjected to DSC and XRD to identify drug excipient interaction.
Preparation of SDLF
A novel SDLF containing a combination of SD and self-emulsifying formulation was prepared by mixing (Table 2) an equal quantity of drug loaded self-emulsifying system to get an nevirapine SDLF (NV-SDLF) (Faisal et al., 2013). Adsorbent NeucilUS2 was added to the above formulation to overcome the stickiness and tackiness and to improve flow properties. The prepared solid NV-SDLF was scrapped, pulverized, sieved, and stored in the desiccator.
Characterization of NV-SES, NV-SD, and NV-SDLF
Differential scanning electron microscope
Thermal analysis was carried out on a DSC Q1000 (TA Instruments, UK). Samples were accurately weighed into aluminum pans and sealed. The sample and reference pans were loaded into the sample chamber at ambient temperature, equilibrated to 25°C, and held at this temperature for 5 minutes. The temperature range for the thermogram was 25°C–250°C, and the samples were heated at a rate of 5°C/minute with an applied modulation of ±1°C every 60 seconds. Samples were purged in a stream of dry nitrogen flowing at 50 ml/minute.
X-ray diffraction
Powder XRD was recorded on an X-ray defractometer (RigakuMiniflex 600) consisting of an X-ray tube operated at 600 watts with a fixed tube current of 15 mA and a voltage of 40 kV. Samples were exposed to radiation, over an angular range of 5°–130° (2θ) at a scanning speed of 0.01°–100°/minute. The diffraction patterns were analyzed by Integrated X-Ray Powder Diffraction Software (PDXL) software (version 1.8.0.3).
Surface morphology of NV-SDLF
Surface characteristics of nevirapine, NV-SDLF, and other excipients were assessed using a scanning electron microscope [ZEISS EVO MA18 with Oxford EDS(X-act)]. Magnified images were taken at an excitation voltage of 15–20 kV.
In-vitro dissolution studies of NV-SD and NV-SDLF
In-vitro dissolutions of NV-SD and NV-SDLF were carried out in 900 ml of phosphate buffer pH 6.8 using USP I dissolution apparatus (Electrolab TDT-08L, India). Samples equivalent to 50 mg nevirapine filled in capsules were used for dissolution study at 50 rpm and 37°C ± 0.5°C. Aliquots of samples were withdrawn at a regular time interval of 10 minutes for 180 minutes. The volume pipetted out was replaced with fresh media to maintain sink condition. Samples taken were filtered through a 0.45 µ membrane and then analyzed using a UV spectrophotometer at 282 nm (Chadha et al., 2010; US Food and Drug Administration).
Ex-vivo permeability study of solid NV-SDLF
The permeability study was carried out using chicken intestinal sac (Nipun and Ashraful Islam, 2014) which was collected from a slaughter house. A cleaned intestinal sac of length 3 cm was tied at one end and filled with nevirapine, and the sample was suspended in 1 ml of water. Tying the other end of the sac, we determined the release of the drug using USP I dissolution apparatus containing 900 ml of basic medium 6.8 phosphate buffer at 50 rpm maintained at 37°C ± 0.5°C. Samples were withdrawn at a predetermined interval and each sampling was replaced with a fresh medium. Samples filtered through 0.45 µ were analyzed for drug release by using UV spectrophotometer at 282 nm.
In-vivo pharmacokinetic studies in the animal model
The bioavailability of the prepared solid NV-SDLF was assessed in male Wistar rats (220–250 g) after taking institution ethical clearance. Experiments were conducted as per the Committee for the Purpose of the Control and Supervision on Experiments on Animals guidelines. Before the experiments, animals (n = 4 for each group) were fasted for 12 hours. Each animal was cannulated in the right jugular vein in anesthetic condition. Suspensions of solid NV-SDLF and the pure drug were prepared in 0.5% carboxymethyl cellulose with gentle agitation. A test group of animals was administered with solid NV-SDLF suspension and a control group of animals was administered with the pure drug at a dose of 60 mg/kg body weight with oral gavage (Inugala et al., 2015; Moretton et al., 2014; Usach and Peris, 2011).
Estimation of nevirapine in the blood sample
Nevirapine was extracted from 200 µl plasma by liquid–liquid extraction using ethyl acetate as the extraction solvent. The extracted dried sample was reconstituted with mobile phase, acetonitrile: 0.1% formic acid (80:20 v/v) containing internal standard (METAXALONE) for introduction into Liquid Chromatography with tandem mass spectrometry (LCMS/MS). LC separation was carried out by introducing 10 µl of the reconstituted sample into High-performance liquid chromatography (HPLC) (Prominence LC-20AT, Shimadzhu, Japan) consisting of C-18 column (Agilent, ZORBAX XB-C18, 5 µm, 4.6 × 50 mm) maintained at 40°C. Throughout isolation, the mobile phase flow rate of 0.500 ml/minute was maintained. The eluted components from the column were delivered to API 4000 Triple Quadruple mass spectrometer (SCIEX, USA), to detect ions.
Pharmacokinetics analysis
Non-compartmental pharmacokinetic analysis of plasma data was carried out using WinNonline software. Parameters determined using WinNonline are as follows: half-life (t1/2), area under the curve (AUC) of plasma drug concentration time curve from zero to infinity (AUC0–∞), AUC from time of administration to the last plasma concentration (AUC0–Last), maximum plasma concentration (Cmax), time of maximum concentration (Tmax), elimination rate constant (K), and relative bioavailability (%F).
Statistical analysis
In-vivo pharmacokinetic data expressed as mean ± SE were analyzed for statistical significant differences between the means of the two groups by unpaired Student’s “t”-test using GraphPad Prism Software. The results were considered significant at p < 0.05.
RESULTS AND DISCUSSION
Preparation of drug-loaded SES
SES has gained attention in the delivery of BCS class II drugs in recent years due to its ability to combat solubility issues of poorly soluble drug and other advantages with respect to increased GI permeability, enhanced lymphatic absorption, and inhibiting first-pass metabolism and Pg protein efflux (Gurram et al., 2015, Sandeep et al., 2013). In the present study, the development of NV-SES was initiated by screening the various oils, surfactants, and co-surfactants based on the solubility of the drug in the individual vehicle and miscibility among each other. Based on the solubility studies, oleic acid a long-chain triglyceride was selected as oil, Cremophor RH 40 as surfactants, and transcutol HP as co-surfactants, with the solubility of 21.05, 15, and 12.23 mg/ml, respectively. In the preparation of SES, the combination of surfactant and co-surfactant ratio plays a prominent role in the formation of smaller particles. As per the requirement stated by Pouton, the ideal combination of surfactant and co-surfactant is 3:1 (Colin, 1997). Hence, 3:1 ratio of Cremophor RH40:Transcutol HP (surfactant/co-surfactant mix) were mixed and added to the different percentages of oils to get different ratios of SES. Drug loading was conducted for all the ratios and the emulsifying ability of these ratios was determined by dilution method and observed for %transmittance at 638.2 nm. Among the SES ratios, the ratio containing 30% of oil and 70% of S/Co mixture emulsified within a minute with the %transmittance of 92.2 with drug loading 20 mg/ml was selected as optimized SES. The optimized SES was analyzed for particle size and was found to be 108.2 nm, which indicates microemulsion formation after dilution. Measurements of PDI and ZP confirm uniform size distribution of particles and the stability of the colloidal system. The particles in the dispersion with large potential will tend to repel each other and there will be no tendency for particles to aggregate (Gurram et al., 2015). The ZP of the optimized SES was observed to be ±36.5 mV, which confirmed good stability. In support of the ZP result, the polydispersibility index of optimized SES was found to be 0.291 which is less than 0.3, which indicates uniform size distribution of dispersed particles (Zupan?i? et al., 2016).
Preparation of drug-loaded SDs
SDs are molecular mixtures of hydrophobic drugs in hydrophilic carriers, where the drug release profile is driven by polymer properties. In SD, the drug is dispersed but not fully solubilized in the excipient matrix which facilitates a higher dose loading (Vo et al., 2013). For the present study, gelucire 50/13, belonging to the gelucire family, was selected as a SD carrier because of its ability to enhance the oral bioavailability of the hydrophobic drugs and the ability to self-emulsify on contact with aqueous media forming a fine dispersion, i.e., microemulsion (Damian et al., 2000). A SD of nevirapine with different ratios of gelucire 50/13 was prepared by the melt method. From the in-vitro release study in 6.8 phosphate buffer (Fig. 1), it was observed that as the gelucire ratio increased, nevirapine solubility also enhanced. With regard to the bulk of the final formulation and drug loading, 1:4 drug to gelucire ratio was used for the preparation of a novel lipid delivery system.
Preparation of SDLF
Although SES has number of justifiable advantages in delivering a poorly soluble drug, in our present study, it was not possible to successfully load the target dose of 50 mg nevirapine within the SES pre-concentrate. With the intention of increasing the drug loading, a new formulation has been designed called SDLF which has the combined benefits of SES and SD. The SDLF was prepared by mixing an equal quantity of optimized SES and the SD. The product SDLF was of semisolid consistency which would have the disadvantage of the storage and the stability issues. The use of adsorbent can minimize the stickiness and tackiness problems of lipid formulations. Neuciline US2, a synthetic, amorphous form of magnesium aluminometasilicate, exhibits multiple excipient roles due to its porosity, greater specific area of 300 m2/g, and adsorbing capacity of 3.2 ml/g (Gupta et al., 2001). The literature supports its applications in the lipid formulations as an adsorbent in the preparation of solid SEDDS and as the carrier in the formulation of lipid SD (Gumaste et al., 2013; Gupta et al., 2001; Kazi et al., 2017). Hence, neuciline US2 was added to the NV-SDLF to get solid NV-SDLF with desired solid consistency accompanying good stability and flow property.
Characterization of formulations
Differential scanning electron microscope
A thermogram of the drugs, gelucire 50/13, neucilin U2, NV-SD, and solid NV-SDLF, are shown in Figure 2. A sharp endothermic peak at 251.35°C in Figure 2f and 50.36°C in Figure 2e indicates the melting point of nevirapine and gelucire 50/13. The DSC curve of SD (Fig. 2d) revealed a shallow peak without any sharp endothermic peak of the drug at 251.35°C; this confirmed the change in drug property from crystalline to amorphous form. Also, the presence of the endothermic peak of the carrier in SD substantiated that there is no change in the melting point of the carrier and suggested the formation of a monotectic system (Sinha et al., 2010). DSC of neuciline U2 in Figure 2c showed characteristic endotherms at 91.17°C and 231.72°C. A solid SDLF without drug and solid NV-SDLF exhibited one endothermic peak at 150.55°C and 146.51°C in comparison with neuciline U2 (Fig. 2c). This may be due to the melting of the mixture exhibiting more than one phase, indicating the minor change in the thermal behavior of Neucilin U2. But there is no obvious appearance of drug endothermic peak at 251.35°C which confirms the amorphous nature of the drug in the SDLF.
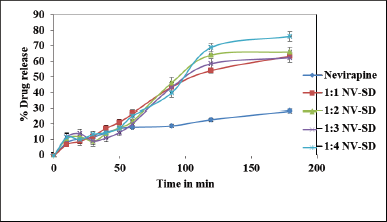 | Figure 1. In vitro drug release of NV-SD in 6.8 phosphate buffer (n = 3). [Click here to view] |
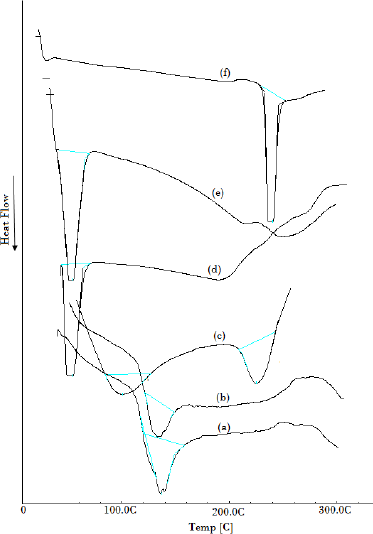 | Figure 2. DSC endothermic peaks: solid SDLF without drug (a), solid NV-SDLF (b), neucilin U2 (c), NV-SD (d), gelucire 50/13 (e), and nevirapine (f). [Click here to view] |
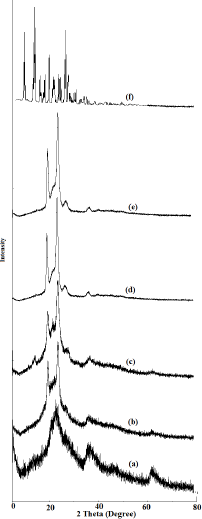 | Figure 3. XRD Peaks: neucilin U2 (a), solid SDLF without drug (b), solid NV-SDLF (c), NV-SD (d), gelucire 50/13 (e), and nevirapine (f). [Click here to view] |
X-ray diffraction
The physical states of nevirapine in NV-SD and solid NV-SDLF were investigated by the X-ray diffraction method and diffractograms are shown in Figure 3. The XRD patterns of pure nevirapine (Fig. 3f) showed distinct peaks between 5° and 30° (2θ = 9.3°, 13.2°, 13.6°, and 25.6°) indicating crystalline nature of nevirapine (Jindal and Mehta, 2015) and diffractogram in Figure 3d shows typical triglycerides peaks at 19.1° and 23.3° of gelucire 50/13 (Chauhan et al., 2005). Triglyceride peaks were retained in NV-SD (Fig. 3d), with suppressed nevirapine crystalline peaks, confirming the transformation of crystalline nevirapine into amorphous nevirapine. As expected, the XRD pattern of neuciline US2 gave no peaks, indicating the amorphous nature of the solidifier. SDLF and N-SDLF retained sharp characteristic peaks of gelucire 50/13 similar to pure gelucire50/13; this result ruled out the possible interaction of drug and gelucire 50/13. The absence of crystalline peaks of nevirapine in solid NV-SDLF reveals that the physical state of the crystalline drug has been transferred into an amorphous form, which is attributed to greater dissolution rate of solid Nevirapine self-dispersing lipid formulation (NV-SDLF) due to higher internal energy (Venkata and Basanth 2013; Tran et al., 2013).
Surface morphology
The surface morphology of the nevirapine, neusilin US2, and solid NV-SDLF was studied by Scanning Electron Microscopy (SEM), as shown in Figure 4. As expected, it was observed that adsorbent neusilin US2 existed as a porous granular spherical fine powder (Fig. 4b), and Figure 4c clearly shows the adsorption of semisolid NV-SDLF on the surface of neusilin US2.
Dissolution studies
Stomach pH can vary due to the presence of food and in fasting conditions (Hamed et al., 2016), and drugs may get absorbed over an extended period of time and reach the small intestine with a physiological pH between 6.5 and 7.6. Therefore, in-vitro dissolution of the formulation NV-SDLF was carried out in the pH, the non-enzymatic simulated gastric pH 0.1 N HCl, and simulated intestinal 6.8 pH phosphate buffer. The release profile of a solid NV-SDLF was compared with the pure drug release profile and it was observed that the drug release in acidic pH was found to be 93% ± 0.91%, which is very close to 99.5% ± 0.84% pure drug release (Fig. 5). In basic pH, solid NV-SDLF exhibited 87.2% ± 0.96% drug release, which was 58.4% more than that of pure drug release (28.8% ± 1.02%). The enhanced drug release in basic pH was attributed to gelucire 50/13, which can enhance drug dissolution (de Oliveira Eloy et al., 2012) and good dispersibility properties of the prepared solid NV-SDLF.
 | Figure 4. SEM images of nevirapine (a), neuciline US2 (b), and NV-SDLF adsorbed on to the surface of neuciline (c) [Click here to view] |
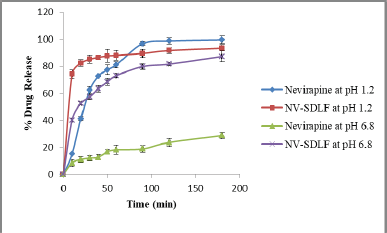 | Figure 5. In vitro drug release of nevirapine solid NV-SDLF in different media (n = 3). [Click here to view] |
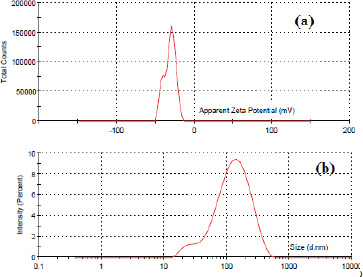 | Figure 6. ZP (a) and particle size (b) of solid NV-SDLF suspension. [Click here to view] |
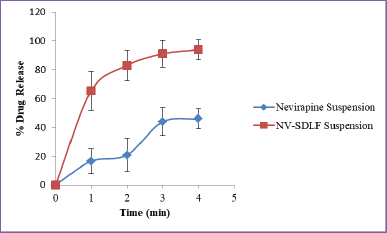 | Figure 7. Ex-vivo drug permeation of nevirapine suspension and NV-SDLF in 6.8 phosphate buffer (n = 3). [Click here to view] |
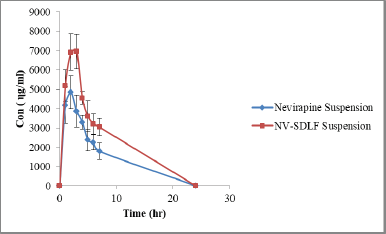 | Figure 8. Plasma concentration time curve of nevirapine suspension and solid NV-SDLF (n = 4). [Click here to view] |
 | Table 2. Composition of NV-SES, NV-SD, and NV SDLF. [Click here to view] |
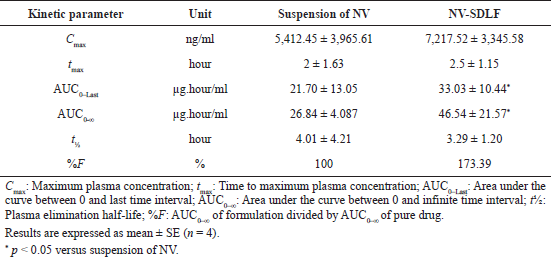 | Table 3. Pharmacokinetic parameters of solid NV-SDLF and suspension of NV. [Click here to view] |
Release kinetics
It was found that the formulated solid NV-SDLF in 0.1N HCl and 6.8 pH phosphate buffer indicated the best agreement with Korsmeyer and Peppas’ release with “n” values of 0.25 (R2 = 0.8072) and 0.080 (R2 = 0.9675), which shows that this release is not governed by the Fickian diffusion. The undefined mechanism of the formulation may be due to the combined effect of SES with SDs.
Particle size and ZP
The in-vivo behavior of the dispersed particle is influenced by particle size. The smaller the size of the particle, the larger will be the surface area, which results in enhanced dissolution and bioavailability (Shweta et al., 2011). As depicted in Figure 6b in the present study the particle size of the dispersed solid NV-SDLF was found to be 100.5 nm, which could justify the enhanced dissolution of the drug in basic pH. Quality of dispersion is assessed by determining PDI. The dispersed NV-SDLF exhibited a polydispersibility index of 0.277, which is less than 0.3 and confirms the good dispersion property of NV-SDLF (Zupan?i? et al., 2016). The stability of dispersed particles depends on the surface charge and its potential; the greater the ZP value of surface charge, the greater will be the repellant forces. The ZP of the NV-SDLF was measured using a zeta sizer, and it exhibited a ZP of ±32.4 mV. The result signifies the stability of the dispersed system (Fig. 6a).
Ex-vivo permeation studies
Nevirapine belongs to BCS class II drug which does not have permeability issues. But for confirming the absence of influence of formulation excipients on permeability, an ex-vivo permeation study was carried out as described by Nipun and Ashraful Islam (2014), using chicken intestinal sac. The permeation profile of the pure drug was compared with solid NV-SDLF and observed that there is no significant difference in drug diffusion profile of the pure drug and optimized formulation (Fig. 7).
Pharmacokinetic studies
The in-vivo oral performance of solid NV-SDLF was compared with nevirapine suspension obtained by dispersing nevirapine in 0.5% w/v carboxymethylcellulose aqueous solution. In this assay, we evaluated the following pharmacokinetic parameters: maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), area under the curve between 0 and last time interval (AUC0–last), area under the curve between zero to ∞ (AUC0–∞), and plasma elimination half-life (t½) using Wistar rats at a dose of 15 mg nevirapine per 1 ml. These parameters of nevirapine were obtained by non-compartmental analysis of plasma concentrations at selected time points. The pharmacokinetic curves of the nevirapine plasma concentration versus time after oral administration are shown in Figure 8. Details of the pharmacokinetic parameters are shown in Table 3. The AUC0–last and AUC0–∞ values observed were 33.03 ± 5.22 and 46.54 ± 21.57 µg/ml for solid NV-SDLF and 21.70 ± 13.05 and 26.84 ± 4.087 µg/ml for nevirapine suspension, respectively. The increase in AUC values was found to be statistically significant at p < 0.05. Also, it was observed that solid NV-SDLF exhibited an increase in Cmax value to 72.17 ± 3,345.58 from 54.12 ± 3,965.61 µg/ml for pure drug suspension, which is nearly 34% increase. The increase in AUC and Cmax was substantiated with increased in-vitro dissolution of Nevirapine. The result is most likely due to the presence of components in NV-SDLF; oleic acid, Cremophor RH 40, and Gelucire, which enhance the bioavailability of the poorly soluble drug by various mechanisms. The expected mechanisms are lymphatic transport of lipophilic drug (long-chain oleic acid) (Zhang et al., 2021), absorption of the lipophilic drug by inhibiting the efflux pumps, cytochrome P450 3A (Cremophor RH 40) (Tayrouz et al., 2003), and solubilizing and emulsifying ability of gelucire (de Oliveira Eloy et al., 2012).
Tmax and t1/2 of nevirapine remained unchanged for both solid NV-SDLF and nevirapine drug suspensions, respectively. This confirmed that the component used in the formulation of SDLF has not interfered in the elimination of the drug and release of the drug from the dosage form.
CONCLUSION
A novel self-dispersing nevirapine formulation was successfully developed by dual formulation approach. Formulated NV-SDLF exhibited enhanced in-vitro dissolution in simulated gastric and intestinal pH and showed good dispersibility properties with desired particle size, PDI, and ZP. NV-SDLF with semisolid consistency was adsorbed onto carrier neuciline US2 to overcome handling issues. Solid NV-SDLF was evaluated for its in-vivo performance in male Wistar rats and characterized by DSC and XRD for drug excipient interaction. A significant increase in AUC was observed for solid NV-SDLF in comparison with pure drug nevirapine, and also solid NV-SDLF showed increased Cmax value. The result proved that added excipient in the formulation of NV-SDLF has the ability to enhance solubility, thereby enhancing bioavailability. At the same time, the added excipients may contribute to the lymphatic transport of the drug and inhibits the efflux pumps and cytochrome P450 3A. Further studies need to be conducted to verify the possibility of converting novel solid NV-SDLF into a suitable dosage form for the effective management of AIDS.
ACKNOWLEDGMENT
Nil.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest to disclose.
FUNDING
None.
AUTHORS’ CONTRIBUTIONS
All authors have made an active contribution in every step of research work and reviewed the final draft before submission to the journal. All the authors are accountable for the information provided in the manuscript.
REFERENCES
Bertoni S, Albertini B, Passerini N. Different BCS Class II Drug-Gelucire Solid Dispersions Prepared by Spray Congealing: Evaluation of Solid State Properties and In Vitro Performances. Pharmaceutics. 2020;12(6):548. CrossRef
Chadha R, Arora P, Saini A, Jain DS. Solvated crystalline forms of nevirapine: thermoanalytical and spectroscopic studies. AAPS Pharm Sci Tech, 2010; 11(3):1328–39. CrossRef
Chauhan B, Shimpi S, Paradkar A. Preparation and characterization of etoricoxib solid dispersions using lipid carriers by spray drying technique. AAPS Pharm Sci Tech, 2005; 6(3):E405–12. CrossRef
Colin W P. Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev, 1997; 25:47–58. CrossRef
Cometa S, Bonifacio MA, Trapani G, Di Gioia S, Dazzi L, De Giglio E, Trapani A. In vitro investigations on dopamine loaded solid lipid nanoparticles. J Pharm Biomed Anal, 2020; 185:113257. CrossRef
da Fonseca Antunes AB, De Geest BG, Vervaet C, Remon JP. Gelucire 44/14 based immediate release formulations for poorly water-soluble drugs. Drug Dev Ind Pharm, 2013; 39(5):791–8. CrossRef
Damian F, Blaton N, Naesens L, Balzarini J, Kinget R, Augustijns P, Van den Mooter G. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur J Pharm Sci, 2000; 10(4):311–22. CrossRef
de Oliveira Eloy J, Saraiva J, de Albuquerque S, Marchetti JM. Solid dispersion of ursolic acid in Gelucire 50/13: a strategy to enhance drug release and trypanocidal activity. AAPS Pharm Sci Tech, 2012; 13(4):1436–45. CrossRef
de Oliveira GG, Ferraz HG, Severino P, Souto EB. Compatibility studies of nevirapine in physical mixtures with excipients for oral HAART. Mater Sci Eng C Mater Biol Appl, 2013; 33(2):596–602. CrossRef
Faisal W, Ruane-O’Hora T, O’Driscoll CM, Griffin BT. A novel lipid-based solid dispersion for enhancing oral bioavailability of Lycopene—in vivo evaluation using a pig model. Int J Pharm, 2013; 453(2):307–14. CrossRef
Gershanik T, Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm, 2000; 50(1):179–88. CrossRef
Gumaste SG, Dalrymple DM, Serajuddin AT. Development of solid SEDDS, V: compaction and drug release properties of tablets prepared by adsorbing lipid-based formulations onto Neusilin® US2. Pharm Res, 2013; 30(12):3186–99. CrossRef
Gupta MK, Goldman D, Bogner RH, Tseng YC. Enhanced drug dissolution and bulk properties of solid dispersions granulated with a surface adsorbent. Pharm Dev Technol, 2001; 6(4):563–72. CrossRef
Gurram AK, Deshpande PB, Kar SS, Nayak UY, Udupa N, Reddy MS. Role of components in the formation of self-microemulsifying drug delivery systems. Indian J Pharm Sci, 2015; 77(3):249–57. CrossRef
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother, 2004; 58(3):173–82. CrossRef
Hamed R, Awadallah A, Sunoqrot S, Tarawneh O, Nazzal S, AlBaraghthi T, Al Sayyad J, Abbas A. pH-Dependent solubility and dissolution behavior of carvedilol—case example of a weakly basic BCS class II drug. AAPS Pharm Sci Tech, 2016; 17(2):418–26. CrossRef
Huang Y, Dai WG. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B, 2014; 4(1):18–25. CrossRef
Inugala S, Eedara BB, Sunkavalli S, Dhurke R, Kandadi P, Jukanti R, Bandari S. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: in vitro and in vivo evaluation. Eur J Pharm Sci, 2015; 74:1–10. CrossRef
Jindal N, Mehta SK. Nevirapine loaded Poloxamer 407/Pluronic P123 mixed micelles: optimization of formulation and in vitro evaluation. Colloids Surf B Biointerfaces, 2015; 129:100–6. CrossRef
Jing-ling T, Jin S, Zhong GH. Self-emulsifying drug delivery systems: strategy for improving oral delivery of poorly soluble drugs. Curr Drug Ther, 2007; 2:85–93. CrossRef
Kazi M, Al-Qarni H, Alanazi FK. Development of oral solid self-emulsifying lipid formulations of risperidone with improved in vitro dissolution and digestion. Eur J Pharm Biopharm, 2017; 114:239–49. CrossRef
Komal P, Jayvadan P, Navin S. Self nano-emulsifying drug delivery system for Embelin: design, characterization and in-vitro studies. Asian J Pharm Sci, 2015; 10:396–404. CrossRef
Krishnamoorthy B, Chellan V R, Natarajan T S, Habibur R SM. Self emulsifying drug delivery system: optimization and its prototype for various compositions of oils, surfactants and co-surfactants. J Pharm Res, 2013; 6:510–4. CrossRef
Kuo YC, Chung JF. Physicochemical properties of nevirapine-loaded solid lipid nanoparticles and nanostructured lipid carriers. Colloids Surf B Biointerfaces, 2011; 83(2):299–306. CrossRef
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm, 2000; 50(1):47–60. CrossRef
Moretton MA, Cohen L, Lepera L, Bernabeu E, Taira C, Höcht C, Chiappetta DA. Enhanced oral bioavailability of nevirapine within micellar nanocarriers compared with Viramune. Colloids Surf B Biointerfaces, 2014; 122:56–65. CrossRef
Nardin I, Köllner S. Successful development of oral SEDDS: screening of excipients from the industrial point of view. Adv Drug Deliv Rev, 2019; 142:128–40. CrossRef
Nipun TS, Ashraful Islam SM. SEDDS of gliclazide: preparation and characterization by in-vitro, ex-vivo and in-vivo techniques. Saudi Pharm J, 2014; 22(4):343–8. CrossRef
Potluri RH, Bandari S, Jukanti R, Veerareddy PR. Solubility enhancement and physicochemical characterization of carvedilol solid dispersion with Gelucire 50/13. Arch Pharm Res, 2011; 34(1):51–7. CrossRef
Sandeep K, Mohanvarma M, Veerabhadhraswamy P. Oral lipid-based drug delivery systems—an overview. Acta Pharm Sin B, 2013; 3:361–72. CrossRef
Shegokar R, Singh KK. Surface modified nevirapine nanosuspensions for viral reservoir targeting: in vitro and in vivo evaluation. Int J Pharm, 2011; 421(2):341–52. CrossRef
Shweta G, Sandip C, Krutika K S. Self-nanoemulsifying drug delivery system for adefovir dipivoxil: design, characterization, in vitro and ex vivo evaluation. Colloids Surf A Physicochem Eng Asp, 2011; 392:145–55. CrossRef
Singh S, Bajpai M, Mishra P. Self-emulsifying drug delivery system (SEDDS): an emerging dosage form to improve the bioavailability of poorly absorbed drugs. Crit Rev Ther Drug Carrier Syst, 2020; 37(4):305–29. CrossRef
Sinha S, Ali M, Baboota S, Ahuja A, Kumar A, Ali J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS Pharm Sci Tech, 2010; 11(2):518–27. CrossRef
Tayrouz Y, Ding R, Burhenne J, Riedel KD, Weiss J, Hoppe-Tichy T, Haefeli WE, Mikus G. Pharmacokinetic and pharmaceutic interaction between digoxin and Cremophor RH40. Clin Pharmacol Ther, 2003; 73(5):397–405. CrossRef
Tran PH, Tran TT, Piao ZZ, Vo TV, Park JB, Lim J, Oh KT, Rhee YS, Lee BJ. Physical properties and in vivo bioavailability in human volunteers of isradipine using controlled release matrix tablet containing self-emulsifying solid dispersion. Int J Pharm, 2013; 450(1–2):79–86. CrossRef
Usach I, Peris JE. Bioavailability of nevirapine in rats after oral and subcutaneous administration, in vivo absorption from gastrointestinal segments and effect of bile on its absorption from duodenum. Int J Pharm, 2011; 419(1–2):186–91. CrossRef
US Food and Drug Administration. Dissolution methods database. FDA. Available via https://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm. (Accessed 20 December 2019).
Vasconcelos T, Marques S, das Neves J, Sarmento B. Amorphous solid dispersions: rational selection of a manufacturing process. Adv Drug Deliv Rev, 2016; 100:85–101. CrossRef
Venkata R K and Basanth B E. A Gelucire 44/14 and labrasol based solid self emulsifying drug delivery system: formulation and evaluation. J Pharm Investig, 2013; 43:185–96. CrossRef
Vo CL, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm, 2013; 85(3 Pt B):799–813. CrossRef
Wang Y, De Clercq E, Li G. Current and emerging non-nucleoside reverse transcriptase inhibitors (NNRTIs) for HIV-1 treatment. Expert Opin Drug Metab Toxicol, 2019; 15(10):813–29. CrossRef
Zhang Z, Lu Y, Qi J, Wu W. An update on oral drug delivery via intestinal lymphatic transport. Acta Pharm Sin B, 2021; 11(8):2449–68. CrossRef
Zupan?i? O, Grieβinger JA, Rohrer J, Pereira de Sousa I, Danninger L, Partenhauser A, Sündermann NE, Laffleur F, Bernkop-Schnürch A. Development, in vitro and in vivo evaluation of a self-emulsifying drug delivery system (SEDDS) for oral enoxaparin administration. Eur J Pharm Biopharm, 2016; 109:113–21. CrossRef