
INTRODUCTION
Oral delivery is considered one of the most widely used and convenient routes for drug administration. Oral absorption of drugs is considered a complicated process that is influenced by many variables, such as unfavorable physicochemical properties of drug, drug absorption site, gastric emptying, and the gastrointestinal transit time (Yin et al., 2013). The stomach and upper small intestine are considered the main site for drug absorption; prolongation of the residence time at or before the site of absorption is considered an effective way to achieve higher drug bioavailability, reduce the frequency of drug administration, and consequently better patient compliance (Kotreka and Adeyeye, 2011; Qin et al., 2018).
Gastroretentive delivery matrix has a great interest especially for the drugs that act locally in the stomach, drugs with narrow absorption window, unstable drugs in the intestinal environments, or drugs that have low solubility at high pH values (Bardonnet et al., 2006). Furthermore, these matrices can be used as a sustained released matrix for drug delivery (Baumgartner et al., 2000; Streubel et al., 2006).
Many approaches have been developed for gastroretentive matrix preparation, as mucoadhesive delivery systems, high-density systems, swelling, and expandable systems and floating systems (Kotreka and Adeyeye, 2011; Lopes et al., 2016; Pawar et al., 2011). Among these, floating systems have been commonly used with minimum adverse effects on the gastric motility and gastric emptying rate (Sathish et al., 2011; Singh and Kim, 2000).
Cefditoren pivoxil is one of the third-generation bactericidal cephalosporins whichthat has a broad-spectrum activity against Gram-positive and Gram-negative bacterial species. It has an inhibitory effect on cell wall synthesis (Barberán and Mensa, 2009; Biedenbach and Jones, 2009; Jones et al., 2000). It is used as frequent treatment for pharyngitis, tonsillitis, acute bronchitis, and skin infection (Alvarez Sala et al., 2006; Balbisi, 2002; Yamada, 2007). During absorption, cefditoren pivoxil is hydrolyzed to cefditoren by esterase enzyme; cefditoren is an active drug moiety that is distributed in systemic blood circulation (Brass et al., 2003). According to Biopharmaceutical Classification System, cefditoren pivoxil is categorized as a System IV compound (low permeability; low solubility) (Chandira et al., 2011; Rumondor et al., 2016). It is a weak lipophilic acid with pka value of 4.2 at 25° (Darkes and Plosker, 2002). Its oral absorption is limited; it is about 16% and increases to 25% with low-fat and high-fat meals. It has a short half-life of 1.6 ± 0.4 hours (Matsumoto et al., 2014).
Our aim in the present study was to investigate the possibility of improving the solubility and enhancing the dissolution rate of cefditoren pivoxil by preparing solid dispersion (SD) using hydrophilic polymers. Additionally, the best formulation will be further selected in floating tablets preparation using hydrophilic or hydrophobic swellable polymers as hydroxypropyl methylcellulose (HPMC) K15M and ethyl acetate by applying 23 full factorial design as an attempt to extend the mean residence time and drug release in the stomach. This will expect to permit a gradual dissolution process and avoid rapid entrance into the small intestine without dissolution, improving the bioavailability of the cefditoren.
MATERIALS AND METHODS
Materials
Cefditoren pivoxil, hydroxypropyl (HPMC KM15), ethyl cellulose, polyvinylpyrrolidone (PVP) (K30), sodium bicarbonate, citric, and Mg stearate were acquired from Sigma Egypt Co. Ltd., Egypt, as a gift sample. Cefditoren Pivoxil capsules (Spectracef®) iswere manufactured by Tedec-Meiji Farma, S.A., Spain). All used reagents were of a high analytical grade.
Preparation of s(SDs) of cefditoren pivoxil
The composition of different SD formulations of cefditoren pivoxil using hydrophilic polymers as PVP and poloxamer 188 at drug-to-carrier weight ratios (1:1, 1:2, and 1:3 w/w) is presented in Table 1. They were prepared using the solvent evaporation technique. Enough volume of dichloromethane was used to dissolve the drug and polymer by applying continuous stirring. Mixing was continued under ambient pressure and temperature in a fume hood until complete solvent evaporation and dry granules were obtained. The dried granules were ground, sieved by 300 mm sieve, and kept in a tightly amber container. Unprocessed drug powder was used as a control.
Preparation of drug physical mixtures
The composition of the physical mixture is presented in Table 1. It was prepared using dry geometric blending of the drug and hydrophilic polymers. The prepared physical mixtures were sieved through a 300 mm sieve and then packed in a closely tight amber container.
Physical characterization of SDs
Differential scanning calorimetry (DSC)
Thermal behavior of cefditoren, PVP, poloxamer 188, and SDs of CSD2 and CSD4 was recorded using a DSC (PerkinElmer Inc., Shelton, CT). This was applied to study the possible compatibilities between cefditoren and chosen polymers. Samples were fastened in an aluminum crimp cell and heated at a speed of 10°C/minutes over a range from 40°C to 300°C using an empty pan as control through the same conditions.
Fourier transform infrared spectroscopy (FT-IR)
FT-IR spectra of cefditoren and SDs (CSD2 and CSD4) were set down using FT-IR spectrometer (Perkin Elmer Life, Shelton, CT). Samples were combined with potassium bromide and then compressed into disks using hydraulic press before scanning. The spectrum of analyzed samples was gathered within the wavenumber region from 4,000 to 350 cm−1.
Determination of dissolution rate of SD mixture
The release of cefditoren pivoxil from SD mixtures was carried out using a USP apparatus I (Copley Dissolution Tester). 900 ml of 0.1 N HCl maintained at 37°C ± 0.5°C was used as dissolution media, with paddle speed of 75 rpm. Samples (5 ml) were taken at 0.0, 5, 10, 15, 30, 45, and 60 minutes, after drug loading (400 mg) or its equivalent amount of the formulations. The sink condition was maintained by replacing the withdrawn sample with the same amount of fresh warm 0.1N HCl. Samples were immediately filtered, and their absorbance was spectrophotometrically determined at 275 nm. The dissolution was evaluated in terms of dissolution efficiency (DE) at 5, 10, and 60 minutes. DE was calculated from the area under the dissolution curve using the trapezoidal rule and determined as a percentage of the area of the rectangle that depicted by 100% dissolution at the same time as shown in Table 1. The resulting values of DE were examined by analysis variance (ANOVA) to find out any statistical difference.
Preparation of floating tablets using 23 full factorial design
Experimental 23 full factorial design
The SD formulations that showed the best release behavior were selected to prepare the floating matrix using 23 randomized full factorial design. The design was devoted to improve the physicochemical characteristics of cefditoren pivoxil floating tablets and to determine the effect of independent variables on times needed for 50% of drug to be released (T50%) and the amount of drug released at 8 hours (Q8) using two levels as shown in Table 2. In the present study, three factors were carefully chosen as independent variables with two levels for every chosen variable, the type of polymer (X1) at two levels (HPMC or ethylcellulose) and the amount of polymer (X2) at two levels (100 or 150 mg) and in the presence or absence of citric acid (X3). The experimental design carried out at all eight possible formulations is shown in Tables 2 and 3. Analysis of variance (ANOVA) was carried out to evaluate model significance, whereas p < 0.05 was considered significant. The composition of the prepared floating tablet matrices (F1–F8) was listed in Table 4.
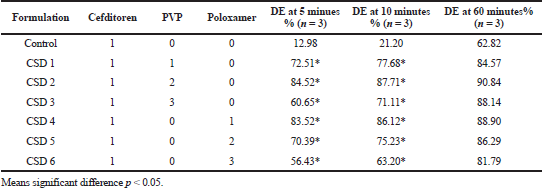 | Table 1. Composition of physical mixture, solid dispersion, and dissolution efficiency DE of cefditoren pivoxil. [Click here to view] |
An equivalent amount of cefditoren pivoxil (400 mg) per tablet was blended with all selected excipients except talc and magnesium stearate after being acutely weighed for 10 minutes using a clean dry mortar and pestle. Magnesium stearate and talc were added to the blend and were mixed continuously for few minutes. Before compression into tablets, the designed amount of each formula was weighted and then compressed using 12 mm punch machine (Erweka tablet press machine, Germany). The compression force of the machine was settled to produce tablets of 4–5 kp in hardness.
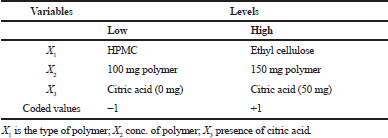 | Table 2. Coded units of 23 full factorial design for cefditoren floating matrices. [Click here to view] |
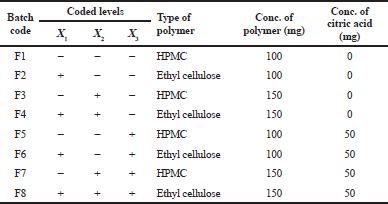 | Table 3. Formulation characteristics of the prepared cefditoren floating tablets. [Click here to view] |
Statistical analysis
Statistical analysis of all parameters was carried out using Minitab statistical software (Minitab release 16.1). The factorial experiment was statistically evaluated by multiple regression analysis. A statistical model associating interactive and polynomial terms was used to express the response shown in equation 1 as follows:
Y = b0 + b1X1 + b2X2 + b3X3 + b12X1X2 + b13X1X3 + b23X2X3 + b123X1X2X3 (1)
where Y is the dependent variable; X1, X2, and X3 are the coded levels of the independent variables; b0 is the arithmetic mean response of the eight runs; and bi is the estimated coefficient for the factor i.
The mean effect (X1, X2, and X3) represents an average of the result of altering one factor at a time from its low to high level. The interaction terms (X1X2, X1X3, X2X3, and X1X2X3) show how the response changes when two factors are simultaneously modified. A negative coefficient indicated the antagonistic relationship, while a positive one reflected the synergistic relationship between factors and response (Gong et al., 2018; Huang et al., 2004).
In vitro evaluation of cefditoren pivoxil floating tablets
Uniformity of weight
Twenty tablets were randomly chosen and accurately weighed. The test was carried out according to USP specification. The tablets meet up with USP specification if no more than two tablets are outer the limit and no tablet varied by more than twice the limit (United States Pharmacopiae National Formulary 38, 2014). Results are represented as an average value ± SD as shown in Table 5.
Tablet thickness
The thickness of ten randomly selected floating matrices was determined using Copley Tablet hardness (Nottingham, UK). Results are recorded as an average value ± SD.
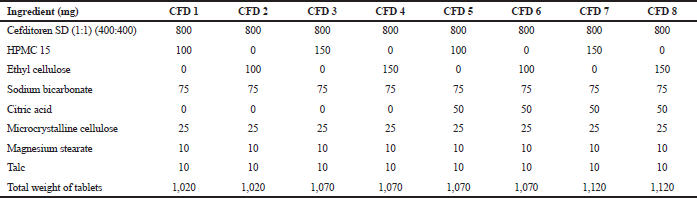 | Table 4. Formulations of cefditoren floating matrices. [Click here to view] |
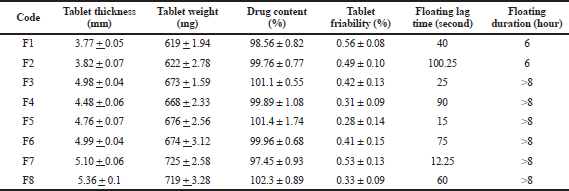 | Table 5. Physicochemical characteristics of cefditoren floating tablets. [Click here to view] |
Tablet friability
The friability of the tablets was determined using Copley Friability Tester (Nottingham, UK). Preweighed, randomly selected 30 tablets were placed and exposed to 100 revolutions. The tablets were pulled out and weighed again. The percentage of loss was calculated, and the value was assumed to be less than 1% (United States Pharmacopiae National Formulary 38, 2014).
Drug content
Ten tablets were randomly chosen, weighed, and crushed. An equivalent quantity to the weight of one tablet was taken out using 100 ml of 0.1N HCl. The resulting solution was carefully filtered through a membrane filter of 0.45 μm pore size. Drug content was measured using UV spectroscopy (Agilent Cary 60 UV-Vis Spectrophotometer, Malaysia) at a wavelength of 276 nm.
Tablet swelling ability
The swelling behavior of the optimized floating matrices (F3) was applied in triplicate, according to the previously designated methods (Dorozynski et al., 2004; Patel et al., 2009). A tablet was accurately weighed (w1), placed in a glass beaker containing 200 ml of 0.1 N HCl, and kept in a water bath at 37°C ± 0.5°C. At a predefined time interval, the tablets were taken away from the beaker and reweighed after the removal of excess liquid. The swollen matrix (w2) was observed over 24 hours. The swelling index (SI) was calculated using eEquation, (2) as follows:
In vitro buoyancy studies
In vitro buoyancy test was carried out for all floating matrices according to the previously published method (Rosa et al., 1994). Briefly, one tablet was placed in 200 ml of 0.1 N HCl and kept up at 37°C ± 0.5°C. The floating lag times and the total floating times are listed in Table 5.
In vitro drug release study of floating tablets
Cefditoren release patterns from reference tablet and floating matrices were evaluated using apparatus II (paddle type) (Copley Dissolution Tester 6000, Nottingham, UK). The dissolution test was implemented using 900 ml of 0.1N HCl, at 37°C ± 0.5°C and 50 rpm for 8 hours. Samples (5 ml) were taken hourly and immediately filtered using a 0.45 μm filter. The test was carried out in triplicate for each matrix. The obtained samples were measured at 276 nm using a UV-Visible spectrophotometer (Agilent Cary 60 UV/Visible Spectrophotometer, Malaysia). The release profile of cefditoren from floating matrices was plotted as the percentage released of cefditoren versus time.
Statistical analysis of drug release profiles of floating tablets
The obtained results were analyzed using Minitab 16.0 software one-way ANOVA. Results were deemed significant if the p-value was less than 0.05.
Kinetic analysis of the release data
To investigate the possible release mechanism of cefditoren from floating matrices, the release results were mathematically analyzed according to the zero-order, first-order, and Higuchi model (Korsmeyer et al., 1983; Liu and Fassihi, 2008; Siber et al., 2005; Tadros, 2010; Zhao et al., 2015). The most appropriate model was selected according to the correlation values (r). The model with the highest (r) was considered the best fitting one (Higuchi, 1963).
Physical characterization of prepared floating tablets
Scanning electron microscopy (SEM)
The surface morphology of optimized floating matrices (F3) was studied 2 hours after tablet dissolution in 0.1 N HCL using SEM (JEOL JSM-6390, Japan).
Physical stability and stability upon storage
Physical stability studies of optimized floating matrices (F3) were carried out according to International Conference on Harmonization guidelines (Mathews, 1999). The optimized formulation was kept in tightly closed containers at ambient conditions (temperature −40°C and relative humidity 75%) for a period of 3 months (Patel et al., 2009). After the predetermined period, the release profile of the tablets was assessed compared to the optimized formulation at time zero and analyzed for any statistical difference.
RESULTS AND DISCUSSION
SDs of cefditoren pivoxil
Solid-state characterization of SDs
The drug content of cefditoren SD was within the acceptable range. Its values were in the range of 98.8%–102.7% w/w. The characterization of solid state was carried out using DSC, FT-IR, and dissolution release studies.
Figure 1 shows the DSC traces of cefditoren in a pure state or SD with selected polymers. DSC showed a single broad peak of pure cefditoren at 130°C corresponding to its melting point. PVP K30 showed a large band of dehydration between 140°C and 160°C, which reflected the amorphous characters of substance and the release of the adsorbed moisture from PVPK30. The endothermic peak of poloxamer 188 appeared at 54°C. This is in good agreement with the reported data for the polymer (El-Habashy et al., 2016). The thermal curves of the selected solid dispersion formula (CSD2 and CSD4) showed the disappearance of the drug characteristic peaks, indicating a change in the morphic form and reduction in drug crystallinity, which would explain the enhanced drug dissolution from SDs. PVP showed a significant disrupting effect on the crystalline structure of cefditoren, especially at medium polymer concentration (1:2).
FT-IR spectroscopy of dispersion prepared powder
The representative FT-IR spectra of cefditoren, PVP K30, and poloxamer 188 in a pure state or in prepared cefditoren SDs are shown in Figure 2. FT-IR spectra of cefditoren and the prepared cefditoren SDs were identical; this referred to the absence of any interaction between cefditoren and the selected polymers used in the investigation of SDs.
Drug dissolution of SD prepared powder
The dissolution release profile of cefditoren in its pure state or cefditoren SDs mixtures with different selected polymers wasis shown in Figure 3. DE values are depicted in Table 1. All SD formulations significantly enhanced drug dissolution compared to the unprocessed drug as shown in Table 1 and Figure 3. The best dissolution parameters were obtained from formulations CSD2 and CSD4 [1:2 drug: PVP; and 1:1 drug: poloxamer (drug to polymer weight ratio), respectively] with an average Q5 (amount release after 5 minutes) of about 85%, compared to only 13% of control; this indicates lipophilicity and/or hydrophobicity of cefditoren. Both formulations showed a DE of about 87% after 10 minutes, compared to only 21% of control. There was a trend of decreased drug dissolution with increasing polymer concentrations, most probably due to increased viscosity in the diffusion layer that retards the solubility of cefditoren pivoxil.
Cefditoren belongs to class IV as reported earlier (Chandira et al., 2011; Rumondor et al., 2016). The main step in drug dissolution mainly depends on the drug ability to dissolve rapidly. Drugs with hydrophobic nature as cefditoren usually have slow, poor solubility that may be attributed to their poor wetting ability. One of the most unpretentious techniques that are usually used to overcome poor solubility problems of drugs is SD method (El Maghraby and El-Sergany, 2014; Betageri and Makarla, 1995). Hydrophilic polymer used in SD preparation may prohibit aggregation of fine particles leading to increase in cefditoren surface area subjected to dissolution media (Venugopala Rao et al., 2014). Furthermore, the formation of SD may originate metastable forms of cefditoren of higher energy, which have the ability of rapid dissolving compared to free cefditoren which can enhance the dissolution rate (Betageri and Makarla, 1995; El Maghraby and El-Sergany, 2014). Based on the mentioned results, the prepared SD of cefditoren with either PVP K30 (1:2, weight ratios) or poloxamer 188 (1:1, weight ratios) can be used to enhance and fasting the solubility of cefditoren. SDs containing PVP K30 (1:2, weight ratios) were chosen to prepare floating matrices according to the composition listed in Table 4.
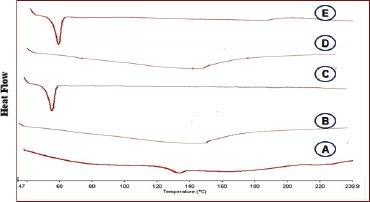 | Figure 1. DSC of cefditoren (A), PVP (B), poloxamer 188 (C), CSF2 (D), and CSD 4 (E). [Click here to view] |
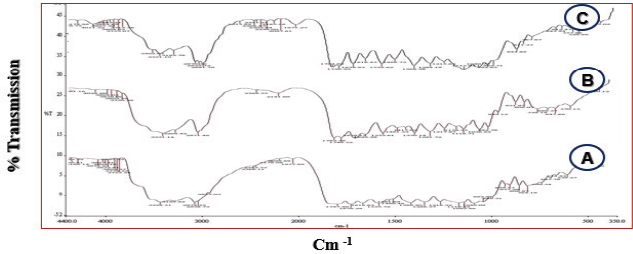 | Figure 2. FT-IR spectra of cefditoren (A), CSD 2 (B), and CSD 4 (C). [Click here to view] |
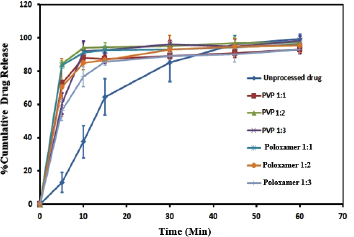 | Figure 3. Release profiles (± SD) of cefditoren from unprocessed drug and drug: polymer ratios (1:1, 1:2, and 1:3 w/w) in 0.1N HCL at 37oC (n = 3). [Click here to view] |
Physicochemical characteristics of cefditoren pivoxil floating tablets
In vitro evaluation of cefditoren pivoxil floating tablets
Floating tablets were developed using hydrophilic polymers such as HPMC K15M or hydrophobic polymer as ethyl cellulose with or without the presence of citric acid.
HPMC K15M was chosen to be a matrixing agent, as it has an extremely good gelling capability in sustained release matrices, as well as being safe, available, and effective (Prajapati et al., 2011). The presence of hydrochloric acid as dissolution medium in addition to sodium bicarbonate is responsible for generating carbon dioxide gas (CO2). The generated CO2 gas is trapped in the gel formed by hydration of HPMC or ethyl cellulose; this causes a decrease in tablet density. The tablet becomes buoyant if the tablet density falls below 1 (Gharti et al., 2012). Ethyl cellulose has the ability to retard drug dissolution, being insoluble in gastric pH (Sakarkar et al., 2010). Incorporating microcrystalline cellulose in the prepared matrices may have led to enhance the swelling ability when the floating matrices come in contact with simulated gastric fluid. This may have led to improved floating capability of matrix by enhancing its water uptake capacity and porosity (Garg and Gupta, 2009).
The hardness of the prepared tablet plays a critical role in affecting its buoyancy by affecting penetration rate of dissolution medium into prepared matrices (Gambhire et al., 2007). Based on these findings, the hardness of the floating matrices was adjusted to be between 5 and 6 kg/cm2, as an attempt to prolong floating lag time of prepared floating matrices.
The physicochemical properties of the prepared floating matrices are listed in Table 5. All the prepared tablet formulations displayed acceptable physicochemical properties according to USP specifications (United States Pharmacopiae National Formulary 38, 2014). The thickness of all floating tablet matrices ranged from 3.77 ± 0.05 to 5.36 ± 0.1 mm. The uniformity of weight was observed in all prepared tablets with less than 1% deviation. This reflected the good flowability properties of the prepared matrices. The observed friability values were in the range of 0.31%–0.56%, which reflected good mechanical resistance of prepared tablets. The range of drug content was between 97.45% and 102.3%, which was within the acceptance criteria of the USP (United States Pharmacopiae National Formulary 38, 2014).
Swelling capability
The swelling ability of the prepared matrices is an important parameter affecting floating properties, capability of adhesion of swellable polymers when coming into contact with gastrointestinal fluid, and consequently the release kinetics pattern of drug from prepared matrix (Tadros, 2010).
The medium uptake by prepared matrices mainly depends on the type of polymer being used as shown in Figures 4 and 5a and b. F7 showed the highest swelling capability during study period. This may be attributed to the high used percent of HPMC K15 M and citric acid related to the test medium. It was clearly noticed that the swelling ability of the matrices increased that by increasing the HPMC K15M concentration.
The swelling attitude of matrices containing HPMC K15M begins with the diffusion of water into the HPMC matrix, forming a concentrated polymer gel. After that, the solvent keeps penetrating into matrix, increasing its dimensions (Gambhire et al., 2007). Alternatively, a significant decrease in the swelling indices (p < 0.05) was noticed with matrices containing ethyl cellulose (F2, F4, F6, and F8). This observed decrease may be attributed to hydrophobic characters of ethyl cellulose causing difficulties in HCl penetration into the matrix (Gambhire et al., 2007; Viridén et al., 2009).
Floating lag time and duration
Gastric floating matrix had to float immediately after its immersion into 0.1 N HCl as dissolution medium and maintain floating for prolonged time (Arora et al., 2005; Dave et al., 2004). In the current study, NaHCO3 was used as a gas-forming agent. During the in vitro test, most of matrices maintained buoyant for more than 8 hours as listed in Table 2. This may be attributed to formation of polymer gel layer with CO2 gas entrapped inside increasing matrix porosity.
The presence of citric acid in fabricated matrix and the acidity of the gastric contents helped in liberation of CO2 gas upon reaching the stomach. This caused a decrease in specific gravity of matrix helping it to float on the chyme (Singh and Kim, 2000). This prolonged residence of drug in stomach might enhance its absorption (Garg and Gupta, 2008).
Floating lag time is another important factor affecting the behavior of the effervescent floating matrices (Table 2). It was observed that the floating lag time of matrices (F1, F3, F5, and F7) was obviously affected by the presence of HPMC KM15 compared to ethyl cellulose. Furthermore, citric acid has significant additional effect on decreasing lag time as shown in formulae F5 and F7 compared to other formulations. This finding might be attributed to the faster penetration of HCl into matrices containing HPMC KM15 and citric acid compared to matrices containing ethyl cellulose.
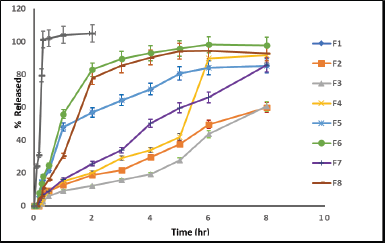 | Figure 4. Release profiles (± SD) of cefditoren formulations (F1–F8) and reference drug in 0.1N HCL at 37oC (n = 3). [Click here to view] |
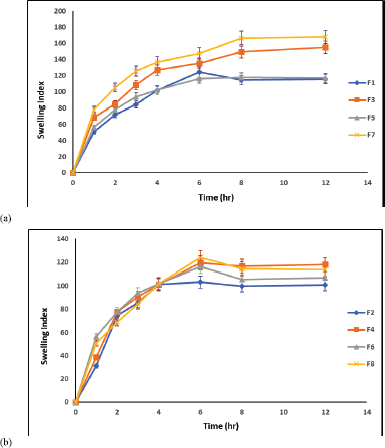 | Figure 5. Swelling indices of cefditoren ?oating matrices using (a) HPMC and (b) using ethyl cellulose in different ratios. Mean ± SD (n = 3). [Click here to view] |
Drug release studies of cefditoren floating tablets
The variety in the selection of polymer(s) type and their applied concentrations resulted in a variety of drug release pattern as shown in Figure 4 and Table 6. The cumulative percentage of drug release for factorial design matrices F1–F8 for 8 hours was found to be in the range of 59.87–97.78%.
F1, F2, and F3 displayed a drug release percent from 59.87% to 69.27% after 8 hours of dissolution. On the other hand, the percent of drug released increased in F4, F5, F6, F7, and F8 to be ranged from 85.23% to 97.78% after 8 hours of dissolution. The results showed that F3 with the highest HPMC concentration and no citric acid content showed the lowest drug release percent compared to the other matrices.
The obtained responses from the design matrix (T50% and % release at 8 hours) showed significant variation (i.e. 1.08–7.18 hours and 59.8%–97.7%, respectively) as indicated in Table 6. F7 containing HPMC K15M (150 mg) and citric acid (50 mg) showed promising dissolution results (t50% = 4.723 hours and Q8 = 85.79%) with good floating behavior. The obtained results reflected the slower release rate for matrices containing HPMC k15M compared with matrices containing ethyl cellulose.
The in vitro dissolution results of cefditoren floating matrices were fitted to linear regression analysis according to zero-order, first-order, and Higuchi’s kinetic equations to specify the mechanism responsible for drug release (Khan and Meidan, 2007). From the above data, F1–F4 and F7 followed zero-order release kinetics (r values ranged from 0.94 to 0.99). F6 and F8 followed first-order release kinetics (r values ranged from 0.97 to 0.98). Only formulation F5 followed Higuchi’s order release kinetics with (r) value of 0.97. Optimization had been carried out using 23 full factorial design to define the effect of applying different amount of HPMC KM15 (X1) and different amount of ethyl cellulose (X2) on the selected independent variables t50% and Q8.
Eqs. (3) and (4) for T50% and Q8 are developed as follows:
T50% = 4.23 − 0.84 X1 + 0.11 X2 − 2.09 X3 − 0.76 X1X2 − 0.16 X1X3 + 0.69 X2X3−0.004 X1X2X3 (3)
% release at 8 hours = 80.4 + 5.16 X1 + 2.33 X2 + 10.0 X3 + 4.37 X1X2 − 0.26 X1X3 − 3.42 X2X3 is 5.75 X1X2X3 (4),
These equations revealed that T50% and % release at 8 hours were markedly dependent on the selected independent variables. The polynomial equations were used to find out the best formulation after estimation of the magnitude of coefficients and the mathematical signs (positive or negative). Plotting the type of polymer (X1), amount of polymer (X2), and the presence or absence of citric acid (X3) versus T50% and % release at 8 hours, respectively, are shown in Figures 6 and 7. Results elucidated that X1, X2, and X3 had a great effect on drug release (T50% and % release at 8 hours). The results indicated that factor X3 (presence or absence of citric acid) has the largest significant (p < 0.05) effect on both responses T50% and Q8, followed by factor X1 (type of polymer) as indicated by their coefficient values. As conclusion, increasing the concentration of HPMC polymer and absence of citric acid favored the sustained release pattern of cefditoren floating matrices. Furthermore, increasing the value of X1 and X2 coefficients proposed the significant effect on T50% due to the interaction between X1 and X2. Consequently, changing in both type and concentration of polymer had a significant effect on cefditoren release rate.
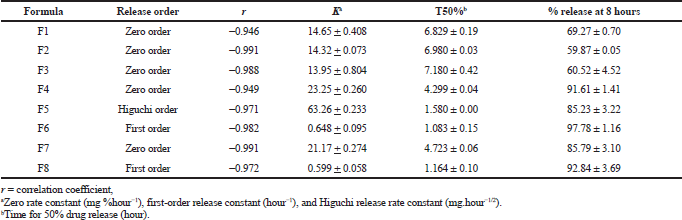 | Table 6. Drug release kinetic parameters and dissolution characteristics of cefditoren floating formulations in 23 factorial designs [mean ± SD (n = 3)]. [Click here to view] |
SEM
Examination of surface morphology of the optimized formulation (F3) was carried out using SEM after 2 hours of tablet dissolution in 0.1 N HCL as shown in Figure 8. The SEM examination revealed the slow entrance of dissolution media into matrix till reaching tablet center forming a swollen network of polymer inside matrix leading to the slow diffusion of cefditoren to surrounding medium.
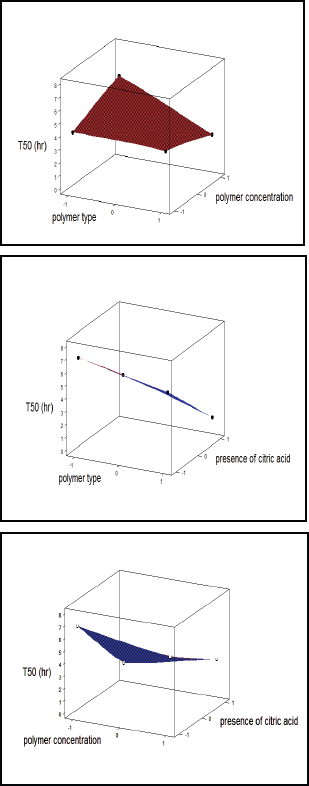 | Figure 6. Surface plot for the effect of independent variables on T50. [Click here to view] |
Physical stability studies and storage stability
Results of stability studies of the optimized formulation (F3) before and after storage are presented in Table 7. F3 was observed to be physically and chemically stable, after 3 months of accelerated stability storage conditions (40°C and 75% RH). Additionally, there was no significant change in release profile of F3 after storage compared to optimized formulation at time zero.
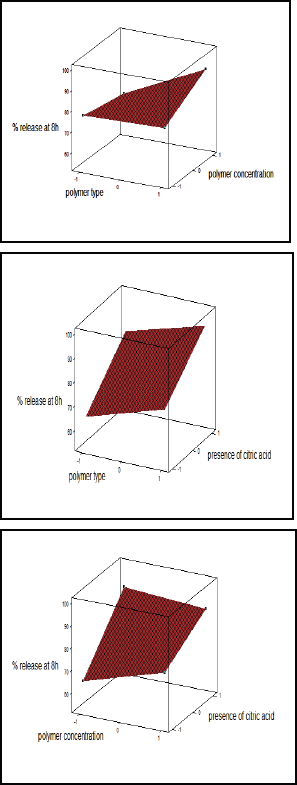 | Figure 7. Surface plot for the effect of independent variables on (Q8). [Click here to view] |
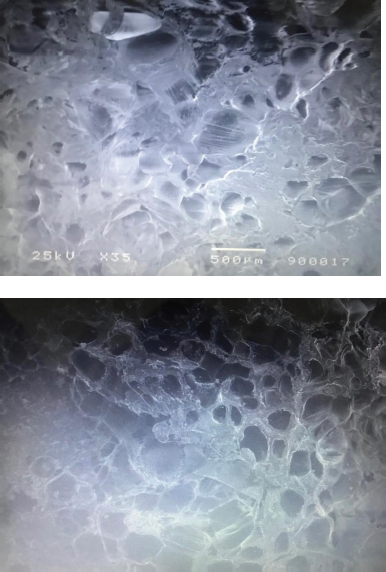 | Figure 8. SEM pictures of optimized F3 after 2 hours in 0.1 N HCL. [Click here to view] |
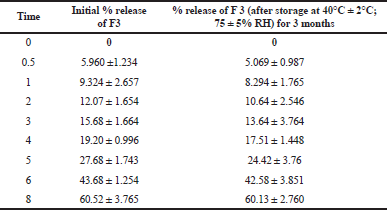 | Table 7. Accelerated stability study of optimized cefditoren floating matrix (F3). [Click here to view] |
CONCLUSION
Cefditoren-sustained release floating matrices were developed and optimized using a 23 factorial design. The floating ability of matrix is due to the generation of CO2 and its entrapment by polymeric matrix. Drug release pattern of the investigated floating tablets was affected by the type of the polymers (HPMC KM15 or ethyl cellulose), the amount of polymer inside matrix (100 or 150 mg), and presence or absence of citric acid. According to the promising in vitro results, it can be deduced that the investigated gastroretentive floating matrices of cefditoren could represent a good alternative of the conventional cefditoren dosage form. However, further clinical experiments should be carried out with the optimized formulation to realize a correlation between in vivo performance and in vitro behavior.
CONFLICT OF INTEREST
There was no conflict of interest declared by authors.
FUNDING
None.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Alvarez Sala JL, Kardos P, Beltran MJ, Coronel P, Group CAW. Clinical and bacteriological efficacy in treatment of acute exacerbations of chronic bronchitis with cefditoren pivoxil versus cefuroxime axetil. Antimicrob Agents Chemother, 2006; 50:1762–7. CrossRef
Arora S, Ali J, Ahuja A, Khar RK, Baboota S. Floating drug delivery systems: a review. AAPS PharmSciTech, 2005; 6:E372–90. CrossRef
Balbisi EA. Cefditoren, a new aminothiazolyl cephalosporin. Pharmacotherapy, 2002; 22:1278–92. CrossRef
Barberán J, Mensa J. Cefditoren and community-acquired lower respiratory tract infections. Rev Esp Quimioter, 2009; 22:144–50.
Bardonnet PL, Faivre V, Pugh WJ, Piffaretti JC, Falson F. Gastro-retentive dosage forms: overview and special case of Helicobacter pylori. J Control Release, 2006; 111:1–18. CrossRef
Baumgartner S, Kristl J, Vrecer F, Vodopivec P, Zorko B. Optimization of floating matrix table and evaluation of their gastric residence time. Int J Pharm, 2000; 195(1–2):125–35. CrossRef
Betageri G, Makarla K. Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques. Int J Pharm, 1995; 126:155–60. CrossRef
Biedenbach DJ, Jones RN. Update of cefditoren activity tested against community-acquired pathogens associated with infections of the respiratory tract and skin and skin structures, including recent pharmacodynamic considerations. Diagn Microbiol Infect Dis, 2009; 64:202–12. CrossRef
Brass EP, Mayer MD, Mulford DJ, Stickler TK, Hoppel CL. Impact on carnitine homeostasis of short-term treatment with the pivalate prodrug cefditoren pivoxil. Clin Pharmacol Ther, 2003; 73:338–47. CrossRef
Chandira RM, Chakraborty BL, Bhowmik D, Goswami S. Formulation evaluation and characterization of cefditoren tablets. Int J Drug Targ, 2011; 2(2):117–25.
Darkes MJM, Plosker GL. Cefditoren pivoxil. Drugs, 2002; 62(2):319–36. CrossRef
Dave BS, Amin AF, Patel MM. Gastroretentive drug delivery system of ranitidine hydrochloride: formulation and in vitro evaluation. AAPS PharmSciTech, 2004; 5(2):77–82. CrossRef
Dorozynski P, Jachowicz R, Kulinowski P, Kwiecinski S, Szybinski K, Skorka, Jasinski TA. The polymers for the preparation of hydrodynamically balanced systems – methods of evaluation. Drug Dev Ind Pharm, 2004; 30(9):947–57. CrossRef
El Maghraby GM, Elsergany RN. Fast disintegrating tablets of nisoldipine for intra-oral administration. Pharm Dev Technol, 2014; 19(6):641–50. CrossRef
El-Habashy SE, Allam AN, El-Kamel AH. Ethyl cellulose nanoparticles as a platform to decrease ulcerogenic potential of piroxicam: formulation and in vitro/in vivo evaluation. Int J Nanomedicine, 2016; 11:2369–80. CrossRef
Gambhire MN, Ambade KW, Kurmi SD, Kadam VJ, Jadhav KR. Development and in vitro evaluation of an oral floating matrix tablet formulation of diltiazem hydrochloride. AAPS PharmSciTech, 2007; 8(3):E73. CrossRef
Garg R, Gupta GD. Preparation and evaluation of gastroretentive floating tablets of silymarin. Chem Pharm Bull, 2009; 57(6):545–9. CrossRef
Garg R, Gupta GD. Progress in controlled gastroretentive delivery systems. Trop J Pharm Res, 2008; 7(3):1055–66. CrossRef
Gharti K, Thapa P, Budhathoki U, Bhargava A. Formulation and in vitro evaluation of floating tablets of hydroxypropyl methylcellulose and polyethylene oxide using ranitidine hydrochloride as a model drug. J Young Pharm, 2012; 4(4):201–8. CrossRef
Gong L, Yu M, Sun Y, Gao Y, An T, Zou M, Cheng G. Design and optimization of gastric floating sustained-release mini-tablets of alfuzosin hydrochloride based on a factorial design: in vitro/in vivo evaluation. Drug Dev Ind Pharm, 2018; 44(12):1990–9. CrossRef
Higuchi T. Mechanism of sustained action medication. J Pharm Sci, 1963; 52:1145–9. CrossRef
Huang YB, Tsai YH, Yang WC, Chang JS, Wu PC, Takayama K. Once-daily propranolol extended-release tablet dosage form: formulation design and in vitro/in vivo investigation. Eur J Pharm Biopharm, 2004; 58(3):607–14. CrossRef
Jones RN, Biedenbach DJ, Johnson DM. Cefditoren activity against nearly 1000 non-fastidious bacterial isolates and the development of in vitro susceptibility test methods. Diagn Microbiol Infect Dis, 2000; 37:143–6. CrossRef
Khan GM, Meidan VK. Drug release kinetics from tablet matrices based upon ethylcellulose ether-derivatives: a comparison between different formulations. Drug Dev Ind Pharm, 2007; 33:627–39. CrossRef
Korsmeyer RW, Gurny R, Docler E, Buri P, Peppas NA. Mechanism of solute release from porous hydrophilic polymers. Int J Pharm, 1983; 15:25–35. CrossRef
Kotreka UK, Adeyeye MC. Gastroretentive floating drug-delivery systems: a critical review. Crit Rev Ther Drug Carrier Syst, 2011; 28:47–99. CrossRef
Liu Q, Fassihi R. Zero-order delivery of a highly soluble, low dose drug alfuzosin hydrochloride via gastro-retentive system. Int J Pharm, 2008; 348(1–2):27–34. CrossRef
Lopes CM, Bettencourt C, Rossi A, Buttini F, Barata P. Overview on gastroretentive drug delivery systems for improving drug bioavailability. Int J Pharm, 2016; 510(1):144–58. CrossRef
Mathews BR. Regulatory aspects of stability testing in Europe. Drug Dev Ind Pharm, 1999; 25:831–56. CrossRef
Matsumoto K, Sato N, Mitomi N, Shitara Y, Shibasaki S. Population pharmacokinetics of cefditoren pivoxil in non-infected adults. Jpn J Antibiot, 2014; 67(1):49–66.
Patel A, Modasiya M, Shah D, Patel V. Development and in vivo floating behavior of verapamil HCl intragastric floating tablets. AAPS PharmSciTech, 2009; 10(1):310–5. CrossRef
Pawar VK, Kansal S, Garg G, Awasthi R, Singodia D, Kulkarni GT. Gastroretentive dosage forms: a review with special emphasis on floating drug delivery systems. Drug Deliv, 2011; 18:97–110. CrossRef
Prajapati S, Patel L, Patel C. Floating matrix tablets of domperidone formulation and optimization using simplex lattice desin. Iran J Pharm Res, 2011; 10(3):447–55.
Qin C, Wu M, Xu S, Wang X, Shi W, Dong Y, Yang L, He W, Han X, Yin L. Design and optimization of gastro-floating sustained-release tablet of pregabalin: in vitro and in vivo evaluation. Int J Pharm, 2018; 545(1–2):37–44. CrossRef
Rosa M, Zia H, Rhodes T. Dosing and testing in-vitro of a bio-adhesive and floating drug delivery system for oral application. Int J Pharm, 1994; 105: 65–70. CrossRef
Rumondor AC, Dhareshwar SS, Kesisoglou F. Amorphous solid dispersions or prodrugs: complementary strategies to increase drug absorption. J Pharm Sci, 2016; 105(9):2498–508. CrossRef
Sakarkar DM, Kshirsagar RV. Deshbhratar RM. Studies on formulation and in vitro evaluation of gastroretentive drug delivery system of carbamazepine. Int J Chem Tech Res, 2010; 2(1):108–13.
Sathish D, Himabindu S, Kumar YS, Shayeda, Rao YM. Floating drug delivery systems for prolonging gastric residence time: a review. Curr Drug Deliv, 2011; 8(5):494–510. CrossRef
Siber BM, Bialer M, Yacobi A. Controlled drug delivery fundamentals and applications. In: Robinson JR, Lee VHL (ed.). 22nd edition, Marcel Dekker Inc, New York, NY, 2005; pp 213–51.
Singh BM, Kim KH. Floating drug delivery systems: an approach to oral controlled drug delivery via gastric retention. J Control Release, 2000; 63:235–59. CrossRef
Streubel A, Siepmann J, Bodmeier R. Drug delivery to the upper small intestine window using gastroretentive technologies. Curr Opin Pharmacol, 2006; 6(5):501–8. CrossRef
Tadros M. Controlled-release effervescent floating matrix tablets of ciprofloxacin hydrochloride: development, optimization and in vitro–in vivo evaluation in healthy human volunteers. Eur J Pharm Biopharm, 2010; 74(2010):332–9. CrossRef
United States Pharmacopiae National Formulary 38. United States Pharmacopial Convention, Rockville, MD, 2014.
Venugopala Rao G, Gadamsetty G, Sarada NC. Formulation and in vitro evaluation of gastro retentive oral hydrogel tablets containing cefditoren. Asian J Chem, 2014; 26(9):2614–6. CrossRef
Viridén A, Wittgren B, Larsson A. Investigation of critical polymer properties for polymer release and swelling of HPMC matrix tablets. Eur J Pharm Sci, 2009; 36(2–3):297–309. CrossRef
Yamada M. Crystal structure of cefditoren complexed with Streptococcus pneumonia penicillin-binding protein 2X: structural basis for its high antimicrobial activity. Antimicrob Agents Chemother, 2007; 51:3902–7. CrossRef
Yin L, Qin C, Chen K, Zhu C, Cao H, Zhou J, He W, Zhang Q. Gastro-floating tablets of cephalexin: preparation and in vitro/in vivo evaluation. Int J Pharm, 2013; 452(1–2):241–8. CrossRef
Zhao Q, Gao B, Ma L, Lian J, Deng L, Chen J. Innovative intragastric ascaridole floating tablets: development, optimization, and in vitro-in vivo evaluation. Int J Pharm, 2015; 496(2):432–9.. CrossRef