
INTRODUCTION
Captopril (Fig. 1), (2S)-1-[(2S)-2-methyl-3-sulphanylpropanoyl]pyrrolidine-2-carboxylic acid, is approved by the Food and Drug Administration (FDA) for the treatment of hypertension, heart failure, left ventricular dysfunction after myocardial infarction, and diabetic nephropathy (European Directorate for the Quality of Medicines and HealthCare, 2007; Marte and Cassagnol, 2019). It is widely employed in geriatric patients suffering from those diseases. However, captopril is administered two to three times per day due to short elimination half-life (Duchin et al., 1988; Marte and Cassagnol, 2019), causing patient incompliance. Therefore, a sustained--release formulation that releases the active pharmaceutical ingredient at the expected rate and duration is needed to cope with the problem (Ansel et al., 2014). Mucoadhesive tablet form is an alternative of sustained-release form that retains the drug in the stomach for a long period, intensifying their contact (Patil and Talele, 2015).
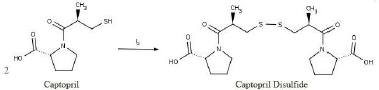 | Figure 1. Reaction of captopril and iodine produces captopril disulfide (Dalla Nora et al., 2019 ). [Click here to view] |
Assay and dissolution are critical parameters in the establishment of oral drug product quality. Assay represents the amount of active pharmaceutical ingredient in dosage form, whereas dissolution illustrates its rate to form a solution. The latter is important in developing drug formulation and in predicting the bioavailability and effectiveness of therapeutics (The United States Pharmacopeial Convention, 2019; Zhang et al., 2010).
Analytical methods to determine the assay and dissolution of captopril immediate-release (IR) tablet are available in the pharmacopeia, but such methods are lacking for captopril mucoadhesive form, which comprises more complex excipients. The United States Pharmacopoeia (USP) utilizes high-performance liquid chromatography (HPLC) for the assay and spectrophotometry for the dissolution of captopril IR tablet, whereas European Pharmacopoeia (EP) employs HPLC in the related substance determination of captopril raw material (European Directorate for the Quality of Medicines and HealthCare, 2007; The United States Pharmacopeial Convention, 2018). Captopril can be transformed into its dimer form, captopril disulfide (CD), which is indicated as the major degradant produced by oxidation (Souza et al., 2012). Therefore, a selective method is required to differentiate captopril from its degradation products. The USP and the EP stipulate good resolution between captopril and CD. However, the EP method is preferred over the USP method because the former uses iodine to produce CD from captopril (Fig. 1), whereas the latter uses a costly CD standard (European Directorate for the Quality of Medicines and HealthCare, 2007; The United States Pharmacopeial Convention, 2018).
In the term of dissolution, the application of a selective method is needed because of possible matrix interference. The results of our preliminary study using the spectrophotometry method as directed in the USP, showed that the matrix of captopril mucoadhesive tablet containing hydroxy propyl methyl cellulose (HPMC), xanthan gum, avicel 102, amylum, magnesium stearate, and talc produces about 70% of interference. Accordingly, a method that can analyze the target component needs to be developed.
Other methods for captopril analysis include flow injection, Fourier Transform Raman spectroscopy, and chemiluminescence (Albero et al., 1993; Fu et al., 2017; Mazurek and Szostak, 2006). However, pharmacopoeial methods are more acceptable for routine quality control compared with non-pharmacopoeial methods. Accordingly, this study aims to adopt a valid method from the USP and the EP for the assay and dissolution of captopril in mucoadhesive tablet form. The methods are validated in accordance with The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Guideline on Validation of Analytical Procedures Q2(R1).
MATERIAL AND METHODS
Material
Captopril working standard was obtained from Phapros, whereas captopril raw material was purchased from Zhejiang Huahai Pharmaceutical Co., Ltd. Some formulas of captopril mucoadhesive tablet which were provided by the Department of Pharmaceutics, Faculty of Pharmacy, Universitas Gadjah Mada, were used in the development of the method, but only two formulas (Formulas A and B) were used in method validation. Formula A consisted of captopril, xanthan gum (Shisam Mas), HPMC K-100 (Colorcon Asia Pasific Pte. Ltd.), avicel 102, amylum, magnesium stearate, and talc (Brataco), whereas Formula B contained captopril, HPMC K-100, ethyl cellulose, talc, amylum, lactose, and magnesium stearate. All of the excipients were pharmaceutical grade.
The reagents used were methanol (HPLC grade, JT. Baker), phosphoric acid, hydrochloric acid, iodine, potassium iodide (p.a., Merck), and aquadest (Brataco), whereas the filter for the mobile phase and the sample were a 0.45 μm cellulose membran filter (Schott Duran) and a 0.45 μm GH Polypro (GHP) syringe membran filter (Milipore Acrodisc GHP). The instruments consisted of HPLC (Hitachi® D2000, pump L2300, detector L2320) equipped with a LiChrospher® 100 RP-8 (250 × 4.6 mm i.d., 5 μm) column, balance (Ohaus PA224, Ohaus EX225D), ultrasonicator (Labocon), micropipette (Gilson), and glassware (Iwaki).
Methods
Assay
Standard preparation
About 25.0 mg of captopril working standard was accurately weighed and transferred into a 100 ml volumetric flask. The standard was dissolved in the mobile phase by a sonicator for 15 minutes. A 2 ml of the solution was transferred into a 10 ml volumetric flask, added with the mobile phase to the mark, and then filtered through a 0.45 μm GHP syringe membrane filter (concentration 50 ppm).
System suitability solution preparation
About 10 mg of captopril was weighed, transferred into a 100 ml volumetric flask, added with a 0.25 ml of 0.05 M iodine and the mobile phase to the mark, and then sonicated for 15 minutes. Iodine was used to oxidize captopril into its dimer form, CD, to test the selectivity of the method. Exactly 1 ml of the solution was transferred into a 10 ml volumetric flask, added with the mobile phase to the mark, and then filtered through a 0.45 μm GHP syringe membrane filter.
Solution A preparation
About 500.0 mg of captopril raw material was accurately weighed, transferred into a 100 ml volumetric flask, added with the mobile phase to the mark, and then sonicated for 15 minutes (concentration 5,000 ppm).
Validation samples preparation
Excipient mixture equivalent to one tablet was weighed, transferred into a 25 ml volumetric flask, added with Solution A (Table 1), added with methanol to the mark, and then sonicated for 15 minutes. Exactly an amount of 0.5 ml of the solution was transferred into a 10 ml volumetric flask, added with the mobile phase to the mark, and then filtered through a 0.45 μm GHP syringe membrane.
The assay method was validated at some parameters: selectivity, accuracy, precision, linearity and range, ruggedness, and robustness. List of the concentration level, replication, and the requirement for each of the parameter are indicated in Table 2.
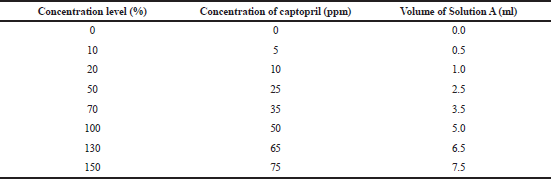 | Table 1. Volume of Solution A at each concentration level of the validation samples of the assay method. [Click here to view] |
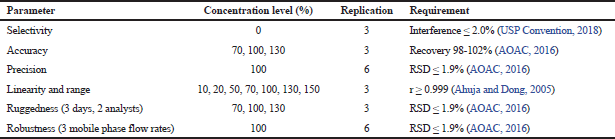 | Table 2. Concentration level applied at each parameter of validation of the assay method. [Click here to view] |
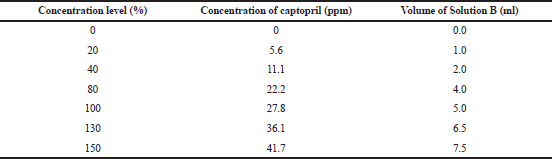 | Table 3. Volume of Solution B at each concentration level of the validation samples of the dissolution method. [Click here to view] |
Dissolution
Standard preparation
About 27.78 mg of captopril working standard was accurately weighed and transferred into a 100 ml volumetric flask. The standard was dissolved in 0.01 N HCl by a sonicator for 10 minutes. One ml of the solution was transferred into a 10 ml volumetric flask, added with 0.01 N HCl to the mark, and then filtered through a 0.45 μm GHP syringe membrane filter (concentration 27.78 ppm). The standard concentration represents the concentration of captopril in 900 ml of 0.01 N HCl if 100% of the captopril in the tablet is completely dissolved.
System suitability solution preparation
A system suitability solution of the dissolution determination method was prepared as directed in the assay, but the final solution was diluted with 0.01 N HCl.
Solution B preparation
About 277.8 mg of captopril raw material was accurately weighed, transferred into a 100 ml volumetric flask, added and dissolved with 0.01 N HCl to the mark, and sonicated for 10 minutes. Ten ml of solution was transferred into a 100 ml volumetric flask, added with 0.01 N HCl to the mark (concentration 277.8 ppm).
Validation sample preparation
Excipient mixture equivalent to one tablet was weighed, transferred into a 100 ml volumetric flask, added with a partial volume of 0.01 N HCl, shaken continuously to avoid agglomeration, and then sonicated for 10 minutes. After the solution reached ambient temperature, it was added with 0.01 N HCl to the mark. Then, 5.0 ml of the solution was transferred into a 50 ml volumetric flask, added with Solution B (Table 3), added with 0.01 N HCl to the mark, and then filtered through a 0.45 μm GHP syringe membrane filter.
The dissolution method was validated in terms of selectivity, accuracy, precision, linearity and range, ruggedness, robustness, and stability of solution. The concentration level, replication, and the requirement for each parameter are listed in Table 4.
HPLC parameter
The assay and dissolution methods were employed below the HPLC parameter. The mobile phase was a mixture of methanol and water containing 0.001% of phosphoric acid (1:1) with a flow rate of 1.0 ml/minute. The injection volume was 20 μl, and the detector was set at 220 nm for assay and at 205 nm for dissolution (European Directorate for the Quality of Medicines and HealthCare, 2007, The United States Pharmacopeial Convention, 2018).
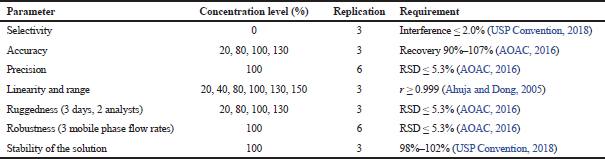 | Table 4. Concentration level applied at each parameter of validation of the dissolution method. [Click here to view] |
Mobile phase preparation
Water containing 0.001% of phosphoric acid was prepared by diluting 85% phosphoric acid with water. An amount of water was placed into a 500 ml volumetric flask, added with 0.5 ml of phosphoric acid 85%, diluted with water to the mark, and then homogenized. The solution was filtered through a 0.45 μm cellulose membrane filter, transferred into a 1000 ml volumetric flask, and then added with methanol to the mark. The mixture was homogenized and degassed for 10 minutes (Agustin, 2019; Damayanti, 2019).
System suitability establishment
The HPLC system was considered ready for the analysis if it can fulfill the following requirements: RSD of the area of captopril obtained from six replicate injections of the standard solution is ≤ 2.0% (The United States Pharmacopeial Convention, 2018), tailing factor of captopril peak is ≤ 2.0 (FDA, 1994), and resolution between the peaks obtained from the system suitability solution is ≥ 2.0 (European Directorate for the Quality of Medicines and HealthCare, 2007).
Calculation
The amount of captopril existing in each sample (%) was calculated as follows:
RESULTS AND DISCUSSIONS
The methods were adopted and modified from the related substance method of captopril raw material in the EP 6.0 and from the dissolution method of captopril IR tablet in the USP 41. The methods were applied to analyze some formulas of captopril mucoadhesive tablet form with a variety of excipients included in the development of the drug. The dissolution method as directed in the USP cannot be used to analyze the formula developed by our laboratory because it produces interference at an intolerant level. However, the chromatographic conditions adopted from the EP can be used for the assay and dissolution methods with some modifications. A problem in the diluent was experienced in the assay sample preparation. When the mobile phase was employed as a diluent for the powdered tablet sample, an agglomerate of the excipient was generated, in which it could not be transferred and filtered. The main possible cause is HPMC, which produces a viscous colloid when added to water (Rowe et al., 2009). As the optimization progressed, methanol was the most suitable diluent because it could dissolve most of the excipients completely and the captopril peak shape was unaffected.
System suitability required the HPLC system to meet the criteria of area RSD and tailing factor of the peak obtained from the standard solution instead of the resolution of the peaks obtained by the system suitability solution. Hence, we injected the standard solution six times and the system suitability solution three times at each run of the analysis. Figure 2 represents the chromatogram, and Table 5 lists a summary of the results. The usage of methanol in the assay and 0.01 N HCl in the dissolution as diluent had no influence on the peak shape, tailing factor, and reproducibility of the area and retention time of captopril. Moreover, the two methods revealed good resolution between captopril and CD, which appeared as the later eluting peak next to captopril because of the low polarity. All of the results met the requirements. On the basis of these results, the methods were validated using some parameters in accordance with the ICH. Formulas A and B were used to validate the dissolution and assay methods, respectively.
Selectivity
Selectivity of the methods was assessed by analyzing the solution prepared from the excipient mixture equivalent to one tablet. The interference was determined by calculating the area obtained in the retention time of captopril by the formula that was previously mentioned. The analysis was performed in triplicate. The excipients used in Formula B produced 0.1% interference in the assay method, whereas the excipients of Formula A generate no interference in the dissolution method. Both methods were considered selective because no more than 2.0% of interference was produced by the excipients (Table 6).
Accuracy
Accuracy of the methods was studied to define the closeness between the true value with the value found in the analysis (ICH, 2005). Accuracy of the assay method was run at three concentration levels of 70%, 100%, and 130%, which covered the expected assay of the tablet. In terms of dissolution, the method was tested for its recovery at four concentration levels of 20%, 80%, 100%, and 130% because the method will be applied to analyze a wide range concentration of captopril due to the release process. Each level was performed in triplicate. The accuracy was evaluated by adding the solution of captopril at a known concentration into the excipient mixture. The assay method was accurate if the recovery was in the range of 98%–102%. The range was determined because the percentage of the active pharmaceutical ingredient (API) to the total weight of the tablet was 10%. For the dissolution, the recovery must be in the range of 90%–107%. Once 100% of the API in a tablet was completely dissolved, the percentage of captopril in 900 ml of 0.01 HCl medium in the dissolution chamber would be 0.002%. The assay and dissolution methods were considered accurate because their recovery rates were 100% and 99%, respectively (Table 7).
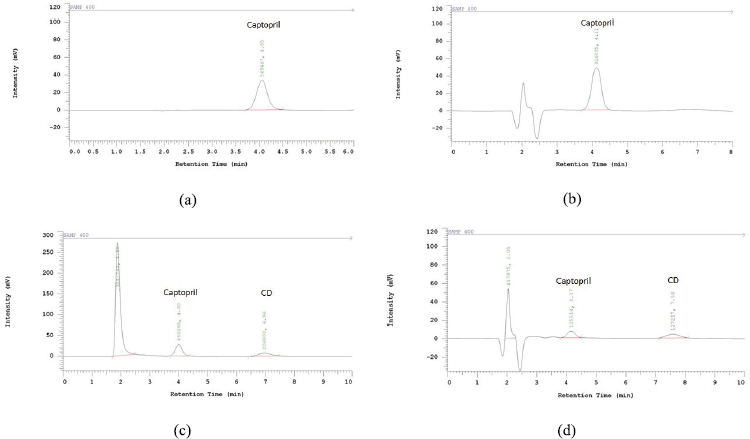 | Figure 2. Chromatogram of standard solution of the assay method (a), standard solution of the dissolution method (b), system suitability solution of the assay method (c), system suitability solution of the dissolution method (d). [Click here to view] |
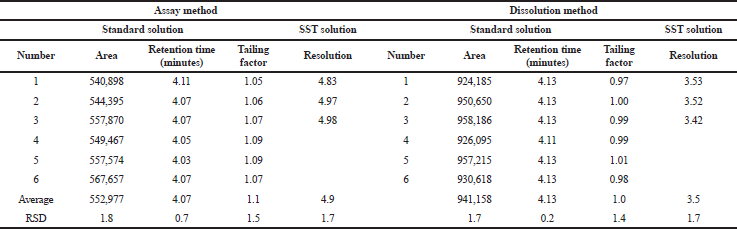 | Table 5. System suitability. [Click here to view] |
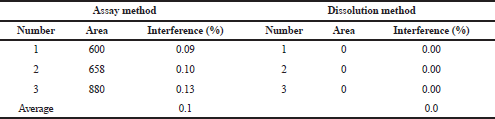 | Table 6. Summary results of the selectivity study. [Click here to view] |
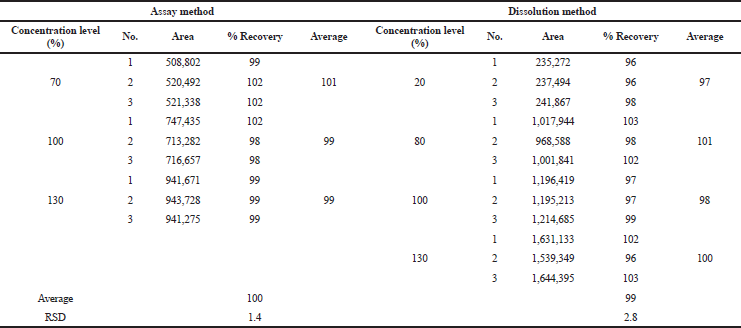 | Table 7. Summary results of the accuracy study. [Click here to view] |
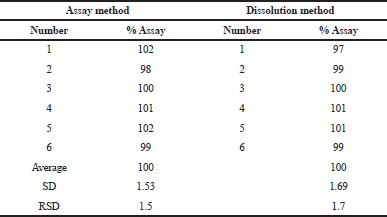 | Table 8. Summary results of the precision study. [Click here to view] |
Precision
The precision of the methods was assessed by analyzing the 100% concentration level of captopril in the excipient mixture six times. The RSD of the assay obtained was then calculated. The assay method was precise if the RSD was not more than 1.9% compared with 5.3% for the dissolution method. The RSD values of the assay and dissolution were 1.5% and 1.7%, consecutively (Table 8).
Linearity and range
Linearity was assessed to determine the ability of the analytical method to obtain a test result that is proportional to the analyte concentration. Furthermore, the range is the interval between the highest and lowest concentration of the analyte in the sample that can be demonstrated by the method in an accurate, precise, and linear way. The linearity was evaluated with a minimum of five levels of concentration, and the range was derived from linearity. The specified minimum range for the assay was between 80% and 120%, whereas that for the dissolution was about 20% over the specification, in which the controlled release dosage form must cover a wide range of specifications (ICH, 2005).
The linearity of the method was determined by creating a linear regression line of the area obtained versus the concentration of the captopril. The line was considered linear if the correlation coefficient was ≥ 0.999. The assay and dissolution methods were linear at the concentration ranges of 5–75 ppm and 5.6–41.7 ppm, respectively. The range of the assay method was 35–65 ppm, whereas that of the dissolution method was 5.6–36.1 ppm (Table 9).
Ruggedness
Ruggedness or intermediate precision was expressed within laboratory variations. This study measures the consistency of the method of delivering the analytical results. The variations can be attributed to the different days, analysts, or instruments (ICH, 2005). The ruggedness of the assay and dissolution methods was established by running an accuracy study in 3 days by two analysts. All of the % recovery values were used to calculate the RSD of the analysis. The requirement of the ruggedness study was the same as that of the precision study. Table 10 infers that both methods are rugged with 1.6% RSD for the assay method and 2.7% RSD for the dissolution method.
Robustness
Robustness represents the capability of the method to maintain its performance during small variations of the method parameter (ICH, 2005). Robustness of the studied methods was carried out by analyzing the sample at 100% concentration level in three mobile phase flow rates (0.8, 1.0, and 1.2 ml/minute). The results of the robustness test are summarized in Tables 11 and 12. The effect of the flow rate on the retention time of the captopril was determined. The higher the flow rate, the shorter the retention time. The resolution of the system suitability solution and the percentage recovery of the captopril were unaffected. The RSD values of the methods were 1.3% and 1.8%. Therefore, the methods were considered robust if changes were observed in the mobile phase flow rate.
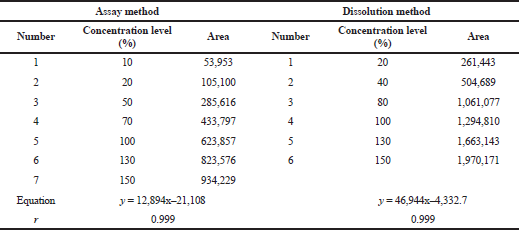 | Table 9. Summary results of the linearity and range study. [Click here to view] |
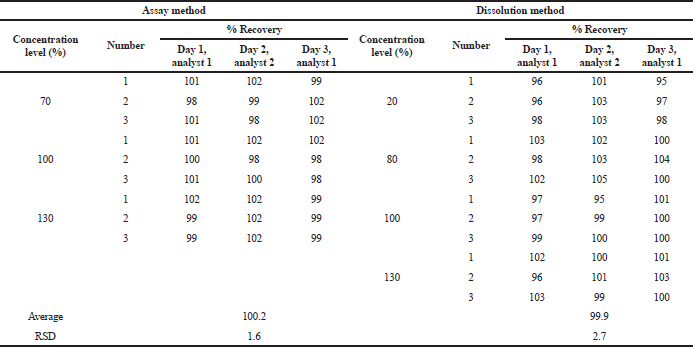 | Table 10. Summary results of the ruggedness study. [Click here to view] |
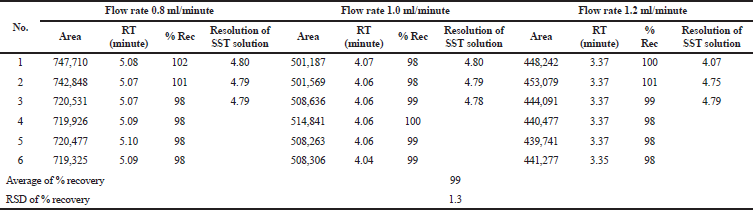 | Table 11. Summary results of the robustness study of the assay method. [Click here to view] |
Stability of solution
This study simulated the idle time before the sample was analyzed after being taken from the dissolution chambers. Stability of the dissolution sample was established by analyzing the sample for 0–300 minutes after preparation. Results showed that the dissolution samples were stable for 300 minutes (Table 13).
The results of the validation study established in accordance with the ICH Guideline met the specification of the assay and dissolution methods in terms of selectivity, accuracy, precision, ruggedness, linearity, and range. Moreover, the methods were successfully applied to analyze some formulas of mucoadhesive tablet form, including formula containing xanthan gum, HPMC K-100, HPMC E6, avicel 102, amylum, magnesium stearate, talc, and ethyl cellulose.
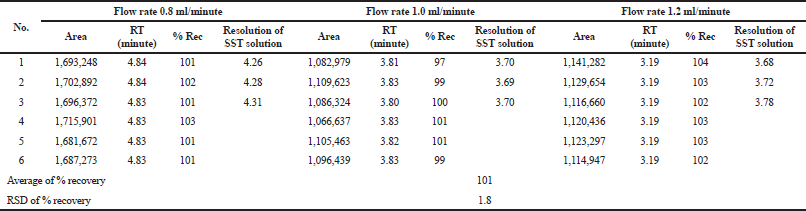 | Table 12. Summary results of the robustness study of the dissolution method. [Click here to view] |
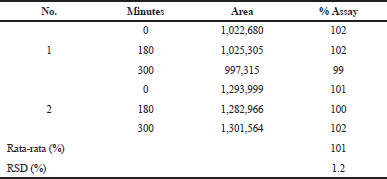 | Table 13. Summary results of the stability study of the dissolution method. [Click here to view] |
CONCLUSION
The assay and dissolution methods of captopril mucoadhesive tablet developed in this research were proven valid in accordance with the ICH Guideline: Analytical Procedure: Text and Methodology. Both methods were also successful in determining the assay and dissolution of some developed formulas of mucoadhesive tablet form. Hence, the methods can be used to analyze captopril mucoadhesive tablets developed in the future.
AUTHORS CONTRIBUTIONS
RBP designed the study and work with EL for data interpretation and drafting of the manuscript. YDA, OD, and ANB were responsible for data acquisition and material preparation.
ACKNOWLEDGMENT
We would like to thank the Dean of the Faculty of Pharmacy, Universitas Gadjah Mada and Directorate of Research Universitas Gadjah Mada for providing funding for this research.
CONFLICT OF INTEREST
All authors declare no conflict of interests in this research.
FUNDING
This research was funded by Faculty of Pharmacy Universitas Gadjah Mada through Hibah Penelitian Dosen Muda in 2018 based on the contract No. UGM/FA/1965.h/M/05/01 and Universitas Gadjah Mada through Kegiatan Peningkatan Kapasitas Peneliti Dosen Muda in 2019 based on the letter of assignment No. 3943/UN1/DITLIT/DIT-LIT/LT/2019.
ETHICAL APPROVALS
This study does not involve the use of animals or human subjects.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Agustin YD. Pengembangan dan validasi metode analisis hasil disolusi tablet sustained release kaptopril secara kromatografi cair kinerja tinggi. Undergraduate thesis. Faculty of Pharmacy Universitas Gadjah Mada, 2019.
Albero MI, Sánchez-Petreño C, García MS, Ródenas V, Determination of captopril in pharmaceutical samples by flow injection analysis. J Pharm Biomed Anal, 1993; 11(10):887-91. CrossRef
Ansel HC, Popovich NG, Allen LV. Pharmaceutical dosage forms and drug delivery systems. 10th edition, Lippincott Williams & Wilkins, Philadelphia, PA, 2014.
AOAC. Guidelines for standard method performance requirements. Association of Official Analytical Chemist Inc., Washington DC, Appendix F, 2016.
Dalla Nora FM, Oliveira AS, Lucas BN, Ferreira DF, Duarte FA, Costa AB, Silva FEB, Barin JS. A novel thermal infrared enthalpimetric method for fast, high-throughput determination of the content uniformity of captopril tablets, J Braz Chem Soc, 2019; 30(5): 1082–8. CrossRef
Damayanti O. Pengembangan dan validasi metode penetapan kadar kaptopril dalam tablet mukoadhesif kaptoril secara kromatografi cair kinerja tinggi. Undergraduate thesis. Faculty of Pharmacy Universitas Gadjah Mada, 2019.
Duchin KL, McKinstry DN, Cohen AI, Migdalof BH. Pharmacokinetics of captopril in healthy subjects and in patients with cardiovascular diseases, Clin Pharmacokinet, 1988; 14(4):241–59. CrossRef
European Directorate for the Quality of Medicines and HealthCare. Captopril monographs, European Pharmacopoeia. 6th edition, Strasbourg, France, 2007.
FDA. Reviewer guidance: validation of chromatographic methods. Center for Drug Evaluation Research, Rocksville, MD, 1994.
Fu Z, Huang W, Li G, Hu Y, A chemiluminescence reagent free method for the determination of captopril in medicine and urine samples by using trivalent silver. J Pharm Anal, 2017; 7(4):252–7. CrossRef
ICH. Validation of analytical procedure: text and methodology Q2(R1). 2005. Available via: https://www.ich.org/page/quality-guidelines (Accessed 24 October 2019).
Marte F, Cassagnol M. Captopril. 2019. Available via https://www.ncbi.nlm.nih.gov/books/NBK535386/ (Accessed 22 October 2019).
Mazurek S, Szostak R. Quantitative determination of captopril and prednisolone in tablets by FT-raman spectroscopy. J Pharm Biomed Anal, 2006; 40(5):1225-30. CrossRef
Patil S, Talele GS. Gastroretentive mucoadhesive tablet of lafutidine for controlled release and enhanced bioavailability. Drug Deliv, 2015; 22(3):312–9. CrossRef
Rowe RC, Sheskey PJ, Queen ME. Handbook of pharmaceutical excipients. 6th edition, Pharmaceutical Press and American Pharmacists Association, Washington, DC, 2009.
Souza JAL, Albuquerque MM, Grangeiro Jr S, Pimentel MF, de Santana DP, SimÕes SS. Quantification of captopril disulfide as a degradation product in captopril tablets using near infrared spectroscopy and chemometrics. Vib Spectrosc, 2012; 62:35–41. CrossRef
The United States Pharmacopeial Convention. Captopril tablets monograph. United States Pharmacopoeia, 41st edition, Rockville, MD, 2018.
The United States Pharmacopeial Convention. Dissolution and drug release tests, 2019. Available via: https://www.usp.org/chemical-medicines/dissolution (Accessed 24 October 2019).
Zhang Y, Huo M, Zhou J, Zou A, Li W, Yao C, Xie S, DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. Am Assoc Pharm Sci J, 2010; 12(3):263-–271. CrossRef