
INTRODUCTION
PIM kinases (PIM-Ks) are significant serine/threonine kinases which are widely used in cellular transcription as well as translation (Cuypers et al., 1984). They occur generally in three distinct isoforms: PIM-1 kinase (PIM-1K), PIM-2K, and PIM-3K. These isoforms have similar functions and are involved in cell processes like multiplication, differentiation, and apoptosis (Fox et al., 2003). These PIM-Ks show weak oncogenic activity in their transgenic forms but exhibit increased activity when expressed along with c-Myc, a transcription factor (Forshell et al., 2011; Zhang et al., 2008). Alteration in the PIM-K pathway is associated with hematological malignancies or solid tumors (Brault et al., 2010). Interestingly, PIM-1 and 2 are progressively expressed in multiple myeloma, lymphomas, and leukemia, whereas PIM-3 is predominantly predicted in the tumors of colon, pancreas, prostate, and some other organelles (Nawijn et al., 2011). Moreover, the deficiency of PIM-1K leads to failure in cell growth and survival. Nevertheless, PIM-1K is linked to drug resistance and is associated with cancer metastasis, immunotherapy, and epigenetic dynamics (Tursynbay et al., 2016). Consequently, PIM-Ks are promising therapeutic targets in drug development for cancer treatment (Asati et al., 2014). The in vivo study on mice proved that the inhibition of these kinases produced minimum side effects (Mikkers et al., 2004). Different inhibitors of PIM-K, for example, SGI-1776, AZD1208, and SMI-4a, were studied against chronic lymphocytic leukemia cells, solid and hematological cancers. However, AZD1208 is under clinical trials (Cervantes-Gomez, et al., 2016; Cortes et al., 2018). In one study carried out, Wang et al. (2015) reported 3-(pyrazin-2-yl)-1H-indazole derivatives as potential PIM-K inhibitors. Furthermore, Xu et al. (2014) have synthesized pyrazolo[1,5-a]pyrimidine derivatives as selective inhibitors of PIM-1K. Keeping the above-mentioned facts in mind, the present study was carried out on a series of pyrazine-linked indazoles to discover the structural requirements as potent PIM-1 inhibitors.
MATERIAL AND METHODS
Data set and Pharmacophore modeling
A data set of 29 compds belonging to 3-(pyrazin-2-yl)-1H-indazoles as inhibitors of PIM-1K were taken for pharmacophore generation, 3D-QSAR, and docking performances. The chemical structures of the above-mentioned series were selected from reported literature (Wang et al., 2015). Different pharmacophore hypotheses were created by using this series, which were subsequently utilized for the 3D-QSAR model creation. The training and test set molecules general selection are given in Table 1. The 3D-QSAR model generation and pharmacophore modeling were carried out by utilizing PHASE module of Schrodinger (Dixon et al., 2006).
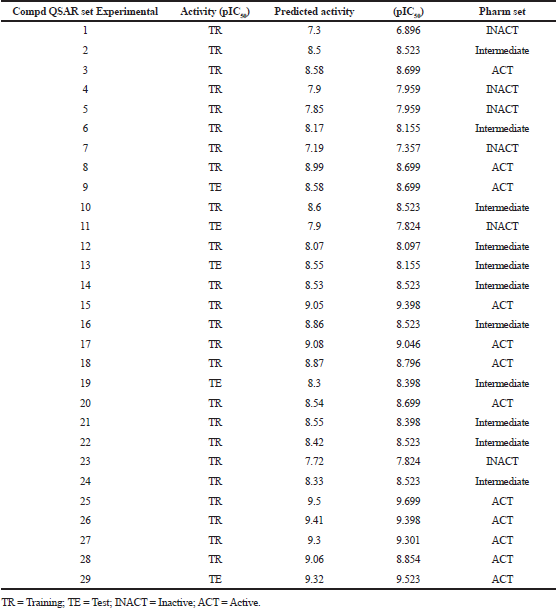 | Table 1. Experimental and predicted activity data for training and test set compds (PLS factors = 5). [Click here to view] |
Ligand preparation
In this module, prepared structures of compds were used and processed for wizard (Ligprep, version 2.5, Schrodinger, NY, 2012). The activity of all molecules was added manually with their pIC50 values. 2D structures were converted into 3D structures through cleanup wizard (Watts et al., 2010). The alignment of all the 29 ligands are shown in Figure 1a. Subsequently, conformer generation was carried out by ConfGen macromodel search method with the application of OPLS-2005 force field (Schrodinger, LLC, 2015). All the molecules were distributed into a training set comprising 24 molecules and a test set of 5 molecules through leave-one-out process. The validation of created models was carried out by utilizing test set. A random activity threshold value was given to split training set molecules into 12 actives (pIC50> 8.6), 11 intermediates (8< pIC50< 8.6), and 6 inactives (pIC50< 8).
Pharmacophore sites Creation and Score hypothesis
The pharmacophoric features were defined by specific SMARTS patterns and common pharmacophores were examined from 12 actives (4.4 version of PHASE, Schrodinger, 2012). Hypotheses were grouped based on the resemblance in identical and variant scores (Table 2). The actives with acceptable fitness scores were carefully chosen and further employed for the 3D-QSAR model creation (Kamaria and Kawathekar, 2014; Sallam et al., 2013).
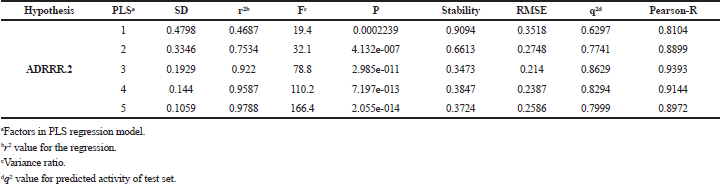
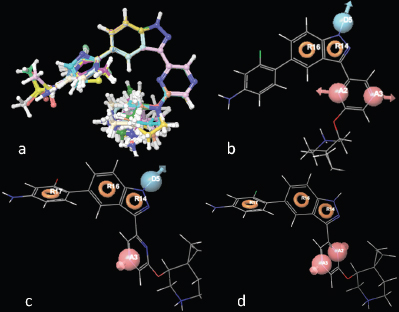 | Figure 1. (a) Alignment of 29 molecules of 3-(pyrazin-2-yl)-1H-indazole derivatives; (b) compd 25 aligned with AADRR; (c) ADRRR; and (d) AARRR. [Click here to view] |
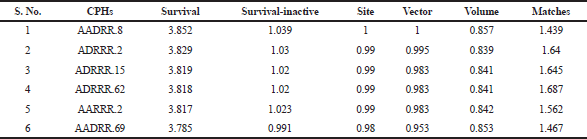 | Table 2. Various hypotheses created by PHASE/scoring results. [Click here to view] |
3D-QSAR data setting
On the basis of aligned ligands of selected hypothesis, the 3D-QSAR models were generated. In the following study, a total of six pharmacophore models were carefully chosen for the final 3D-QSAR models development (Golbraikh and Tropsha, 2002; Lather et al., 2008). These 3D-QSAR models help in elucidation of SAR of all compds (Shah et al., 2010).
Virtual screening
The pharmacophore (ADRRR) used for study and different libraries, like NCI (National Cancer Institute) and ZINC (2.5 million commercially available compds), were used for the virtual screening studies (Irwin and Shoichet, 2005). Lipinski rules of five were applied to choose the drug like compds to develop the potential inhibitors (Lipinski et al., 2001).
Molecular docking
Docking studies on binding site of PIM-1K for 3-(pyrazin-2-yl)-1H-indazoles were accomplished by Glide v3.8 module of Schrodinger, NY (Friesner et al., 2004, 2006; Halgren et al., 2004; Sliwoski et al., 2014). Protein data bank was used to obtain the PIM-1Ks (PDB ID: 2XJ1) crystal structure (Protein preparation wizard Schrodinger, 2012). Another comparative study of 3-(pyrazin-2-yl)-1H-indazoles was carried out on PIM-1Ks crystal structure by Autodock Vina module (Seeliger and Groot, 2010).
MM/GBSA-based rescoring and enrichment calculation
This approach was employed to compute the binding free energy of compds to attain a better estimation of binding powers/strengths and relative potencies for PIM-1K inhibitory activity (Sun et al., 2014). The validation of virtual screening protocols was carried out by computing their enrichment factors as well as receiver operating curve (ROC) analysis (Hamza et al., 2012; Sierra et al., 2015).
ADME analysis
The compds were subjected for their ADME analysis to determine the drug-likeness properties by using QikProp module, version 4.3, Schrodinger, New York, 2015 (Becke, 1993; Lee et al., 1988).
RESULTS AND DISCUSSION
Pharmacophore modeling
In 133 pharmacophore hypotheses of 62 variant, only 6 (AADRR.8, ADRRR.2, ADRRR.15, ADRRR.62, AARRR.2, and AADRR.69) were selected (Table 2) for the development of the final QSAR model. These hypotheses include only three variants with different pharmacophoric features which display best alignment of actives representing top scoring hypotheses. Figure 1b–d shows the alignment of compd 25 (most active) with the highest scoring hypothesis (AADRR, ADRRR, and AARRR) from each of the three variants, respectively. The created hypotheses effectively recognized utmost significant pharmacophoric features suggested for the anticancer potential of 3-(pyrazin-2-yl)-1H-indazoles: A1 containing nitrogen atom, R for aromatic ring, and hydrogen bond donor (HBD) at D5. The deprivation of any of these structural features would compromise the anticancer potential.
3D-QSAR study
To develop a correlation among 3D spatial arrangement of the pharmacophoric features and anticancer activity of 3-(pyrazin-2-yl)-1H-indazoles (Table 3), the best 3D-QSAR model was created. Pharmacophore hypotheses were employed for the alignment of compds to build atom-based 3D-QSAR models through partial least square (PLS) method. The best generated 3D-QSAR model was based on the alignment of ADRRR.2 pharmacophoric features with the most active molecule (Fig. 2a). Table 4 shows the details of the best model of 3D-QSAR including three PLS factor model having excellent statistics as well as predictive capability as depicted from r2 = 0.922, q2 = 0.8629, and Pearson R = 0.9393 values. The model’s validity was expressed by internal predictivity (q2) which can be acquired by leave-one-out (LOO) method. The q2 obtained through LOO method is statistically robust and more reliable as compared to r2 as it is acquired by internal validation method. The high F value (78.8) designates a significant statistical regression model supported by small variance ratio (p = 2.985e-011), which denotes a high level of confidence. Smaller values of SD of regression (0.1929) and RMSE (0.214) provided an apparent suggestion that data which were used to create model in QSAR analysis are the best. A good correlation was obtained among estimated and actual anticancer activity (Table 1). The scatter plots between the estimated versus actual activities displayed that pIC50 values are efficiently speculated for training and test set molecules (Fig. 3a,b). The study results of 3D-QSAR can be seen in Figure 2b–d. Blue blocks present in the pharmacophore model (3D) denote the ligand regions in which particular structural attribute is essential for better activity, while red blocks refer that certain structural attributes are not important for activity. Here compd 25 is the most active inhibitor containing several substituents which affects various features, like HBD, electron-withdrawing, and hydrophobic effects. Introducing HBD and electron-withdrawing groups (EWGs) at ring (R17) lead to increased PIM-1K inhibition. The incorporation of EWGs on pyrazine ring causes decreased inhibition. The presence of hydrophobic groups showed different effects on various positions of these compds in which introduction on R17 is beneficial for activity, but on pyrazine ring the activity depends on ortho, para, and meta substitution.
Molecular docking studies
The active molecule 25 with receptor cavity is shown in Figure 4a. Here, indazole NH group was found engaged in hydrogen bond interaction with Glu121 in the hinge region. The Azaspiro group showed interaction with Asp186 and Glu171, essential for activity. The amine group of phenyl ring showed interaction with Asp131. The extra precision (XP) Gscore of compd 25 was −11.084. All these study data revealed the essential interactions for the ligand with the receptor for better PIM-1K inhibition. Another study for determining the PIM-1K and ligand interaction was established by carrying out molecular docking using the software Autodock Vina. The most active compd 25 binds with different sites on PIM-1K giving information for further structural optimization of active compds. The binding site (Fig. 4b) validates the previous study where different amino acid residues (AARs) like Lys67, Glu171, Glu121, and Asp128 were proven to be essential for activity (Asati et al., 2018). The lowest value of RMSD 2.067 Å as well as binding affinity −9.4 kcal/mol proved better PIM-1K inhibitory activity. The hydrogen bond distance between Glu171, Glu121, and Asp128 to -NH were consecutively 3.3, 3.3, and 2.9 Å. When compd 1 docked with the PIM-1K, it showed only one interaction with Glu121 AAR (Fig. 4c). This interaction has 0.1 Å, the lowest RMSD value with −10.9 kcal/mol binding affinity evidenced least PIM-1K inhibitory activity. The top virtually hit compds, ZINC05885218, ZINC05888770, ZINC08652441, and ZINC73096248 exhibited best XP binding affinity with identical binding interactions to the receptor 2XJ1 evidenced by crystallographic ligand. These compds were showed similar binding attributes with the AARs Glu121, Lys67, Glu171, and Asp128 which are mandatory for PIM-1K inhibition. Three residues of PIM-1K, such as Glu124, Val126, and Ser54, adjacent to ATP-binding site also distinguished PIM-1K from PIM-2K. ZINC05885218 and ZINC05888770, and ZINC08652441 and ZINC73096248 displayed hydrogen bond interaction of -NH group with Glu121, important site for the PIM-1K inhibition (Fig. 5).
MM/GBSA-based rescoring
The Van der Waals (VDWs) interaction was present in docked compds between −33.06 and −54.86 kcal/mol. Similarly, Coulomb energy found in docked complexes fluctuated between positive and negative ranges, where ZINC73096248 showed −103.96 kcal/mol. The complex energy for all compds was found in the negative range between −11,335.46 and −11,403.11 kcal/mol, which evidently indicated that important interactions were VDWs forces. However, the best binding with ΔGbind was found at −55.02 kcal/mol for the top scored (XP docking) compd ZINC73096248 (−7.256 kcal/mol), which also exhibited higher Coulomb energy (−103.96 kcal/mol), and a slightly higher VDWs contribution (−35.76 kcal/mol) with complex energy (−11,368.68 kcal/mol). Henceforth, it is apparent that non-polar VDWs interactions are the principal driving force for binding the ligand in the PIM-1Ks catalytic pocket (Table 5).
Estimation of the virtual screening efficiency
The present study finds the best protocol for the maximum active compd enrichment at the top 5% (6) of the given database. The retrieval of active compds incorporated into the decoy set is presented in Table 6. The actives exhibited enrichment factor with 0.97 ROC and 0.97 AUC.
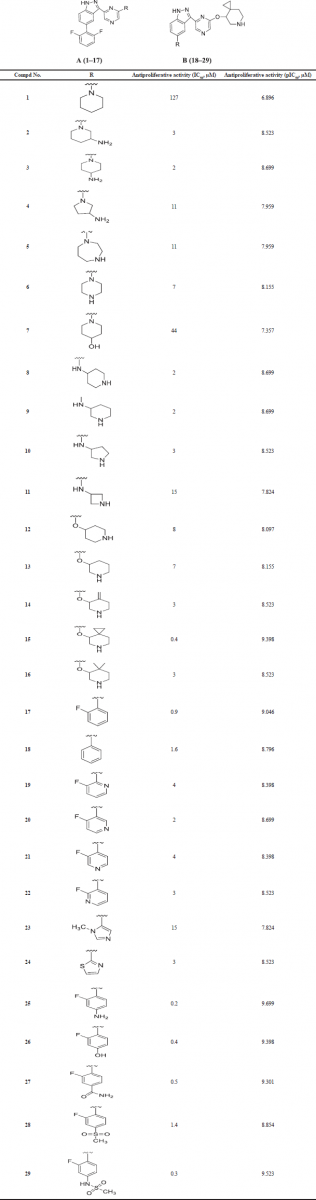 | Table 3. Anti-proliferative potentials of compds 1–29 against PIM-1K. [Click here to view] |
 | Table 4. Statistical parameters of the best atom-based 3D-QSAR model generated by PHASE. [Click here to view] |
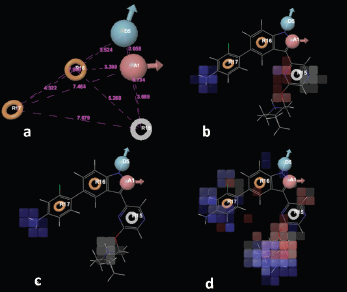 | Figure 2. (a) Common pharmacophoric features of ADRRR.2; (b) the 3D-QSAR model (atom-based) for ADRRR.2; (c) HBD; and (d) hydrophobic effects. [Click here to view] |
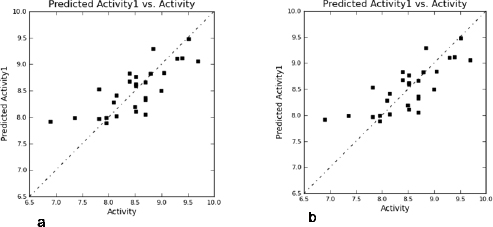 | Figure 3. Scatter plots: (a) training set compd; (b) test set compd. [Click here to view] |
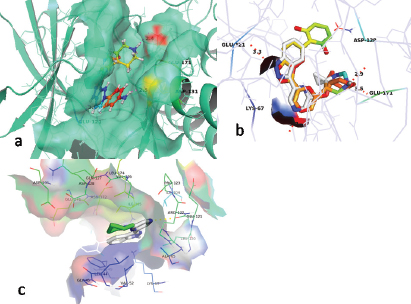 | Figure 4. (a) Binding interaction of compd 25 with the active site of PIM-1K (PDB ID: 2XJ1); (b) comparison of binding interaction of the most active compd 25 and crystal ligand with receptor cavity of PIM-1K (PDB ID:2XJ1); and (c) binding interactions of the least active compd 1 with PIM-1K receptor cavity. [Click here to view] |
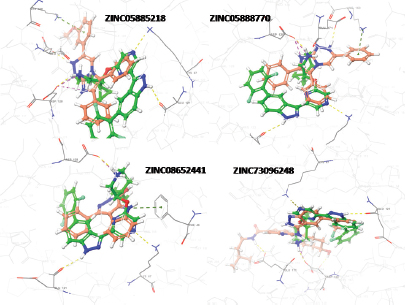 | Figure 5. Hydrogen bond interaction (yellow dotted lines) of compds (red colour) ZINC05885218, ZINC05888770, ZINC08652441, and ZINC73096248, with AARs Asp186, Lys67, and Glu121 of PIM-1K (PDB ID: 2XJ1), respectively. [Click here to view] |
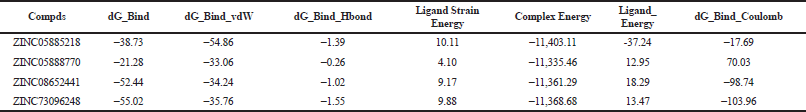 | Table 5. Rescoring of virtually screened compds based on MM/GBSA. [Click here to view] |
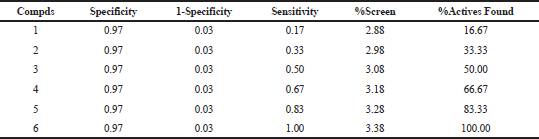 | Table 6. Enrichment factor calculation [Click here to view] |
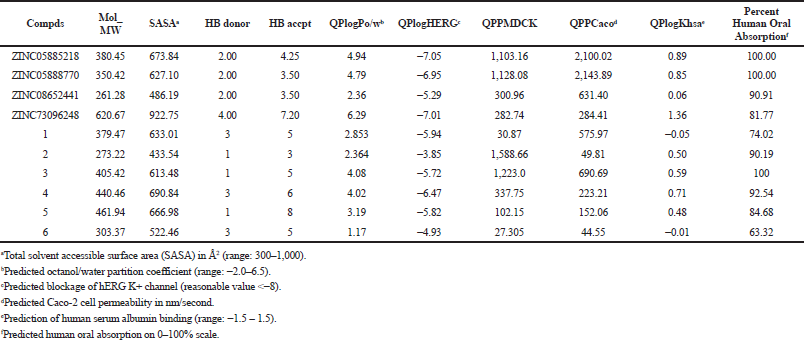 | Table 7. Virtually screened compds (ADME and drug-likeness properties). [Click here to view] |
ADME analysis
The study results showed that pharmaceutical and physicochemical characteristics of all investigated compds were within the acceptable range. The partition coefficient (QPlogPo/w) is a crucial factor for absorption estimation, ranging from 2.36 to 6.29, for the virtually screened compds. QPPCaco (cell permeability), a significant aspect for metabolism of drug, was found between the range of 284.41–2,143.89, and QPPMDCK ranged from 282.74 to 1,128.08. Generally, oral absorption percentage in humans for the screened compds was in the range of 81%–100%. The pharmacokinetic parameters for the screened compds were found within a permissible range (Table 7), thus interpreted as drug-like molecules.
CONCLUSION
The current study highlights the structural requirements of 3-(pyrazin-2-yl)-1H-indazole derivatives for the inhibitory activity against PIM-1K. The study results indicated that the incorporation of strong EWGs into pyrazine ring linked to indazole ring lead to a decreased PIM-1K inhibitory activity. Results of molecular docking of screened compds exhibited identical binding interactions within the PIM-1Ks catalytic pocket in comparison with their corresponding crystal structures. In conclusion, it is inferred that the acquired study results may serve as a guide for designing novel PIM-1K inhibitors, which could be an effective way to discover novel leads for the development of more active as well as safer anticancer agents/drugs.
CONFLICT OF INTEREST
Authors declare no conflict of interest.
FUNDING
None.
REFERENCES
Asati V, Bharti SK. Design, synthesis and molecular modeling studies of novel thiazolidine-2,4-dione derivatives as potential anti-cancer agents. J Mol Struct, 2018; 1154:406–17. CrossRef
Asati V, Mahapatra DK, Bharti SK. Thiazolidine-2,4-diones as multi-targeted scaffold in medicinal chemistry: potential anticancer agents. Eur J Med Chem, 2014; 87:814–33. CrossRef
Becke AD. A new mixing of Hartree-Fock and local density-functional theories. J Chem Phys, 1993; 98:1372–7. CrossRef
Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica, 2010; 95(6):1004–15. CrossRef
Cervantes-Gomez F, Lavergne B, Keating MJ, Wierda WG, Gandhi V. Combination of Pim kinase inhibitors and Bcl-2 antagonists in chronic lymphocytic leukemia cells. Leuk Lymphoma, 2016; 57(2):436–44. CrossRef
Cortes J, Tamura K, DeAngelo DJ, Bono JD, Lorente D, Minden M, Uy GL, Kantarjian H, Chen LS, Gandhi V, Godin R, Keating K, McEachern K, Vishwanathan K, Pease JE, Dean E. Phase I studies of AZD1208, a proviral integration Moloney virus kinase inhibitor in solid and haematological cancers. Br J Cancer, 2018;118(11):1425–33. CrossRef
Cuypers HT, Selten G, Quint W, Zijlstra M, Maandag ER, Boelens W, Wezenbeek PV, Melief C, Berns A. Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell, 1984; 37(1):141–50. CrossRef
Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des, 2006; 20(10–11):647–71. CrossRef
Forshell LP, Li Y, Forshell TZP, Rudelius M, Nilsson L, Keller U, Nilsson J. The direct Myc target PIM3 cooperates with other PIM kinases in supporting viability of Myc-induced B-cell lymphomas. Oncotarget, 2011; 2(6):448–60. CrossRef
Fox CJ, Hammerman PS, Cinalli RM, Master SR, Chodosh LA, Thompson CB. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev, 2003; 17(15):1841–54. CrossRef
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem, 2004; 47(7):1739–49. CrossRef
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem, 2006; 49(21):6177–96. CrossRef
Golbraikh A, Tropsha A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J Comput Aided Mol Des, 2002; 16(5–6):357–69. CrossRef
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem, 2004; 47(7):1750–9. CrossRef
Hamza A, Wei NN, Zhan CG. Ligand-based virtual screening approach using a new scoring function. J Chem Inf Model, 2012; 52(4):963–74. CrossRef
Irwin JJ, Shoichet BK. ZINC-a free database of commercially available compounds for virtual screening. J Chem Inf Model, 2005; 45(1):177–82. CrossRef
Kamaria P, Kawathekar N. Ligand-based 3D-QSAR analysis and virtual screening in exploration of new scaffolds as plasmodium falciparum glutathione reductase inhibitors. Med Chem Res, 2014; 23:25–33. CrossRef
Lather V, Kristam R, Saini JS, Kristam R, Karthikeyan NA, Balaji VN. QSAR models for prediction of glycogen synthase kinase-3b inhibitory activity of indirubin derivatives. QSAR Comb Sci, 2008; 27(6):718–28. CrossRef
Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B, 1988; 37(2):785–9. CrossRef
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev, 2001; 46(1–3):3–26.
Mikkers H, Nawijn M, Allen J, Brouwers C, Verhoeven E, Jonkers J, Berns A. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol, 2004; 24(13):6104–15. CrossRef
Nawijn MC, Alendar A, Berns A. For better or for worse: the role of pim oncogenes in tumorigenesis. Nat Rev Cancer, 2011; 11(1):23–34. CrossRef
Sallam A, Houssen WE, Gissendanner CR, Orabi KY, Foudah AI, Sayed KE. Bioguided discovery and pharmacophore modeling of the mycotoxic indole diterpene alkaloids penitrems as breast cancer proliferation, migration, and invasion inhibitors. Medchemcomm, 2013; 4(10):1360–9. CrossRef
Seeliger D, Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des, 2010; 24(5):417–22. CrossRef
Shah UA, Deokar HS, Kadam SS, Kulkarni VM. Pharmacophore generation and atom-based 3D-QSAR of novel 2-(4-methylsulfonylphenyl)pyrimidines as COX-2 inhibitors. Mol Divers, 2010; 14(3):559–68. CrossRef
Sierra C, Ordonez C, Saavedra A, Gallego JR. Element enrichment factor calculation using grain-size distribution and functional data regression. Chemosphere, 2015; 119:1192–9. CrossRef
Sliwoski G, Kothiwale S, Meiler J, Lowe EW. Computational methods in drug discovery. Pharmacol Rev, 2014; 66(1):334–95. CrossRef
Sun H, Li Y, Shen M, Tian S, Xu L, Pan P, Guan Y, Hou T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys Chem Chem Phys, 2014; 16(40):22035–45. CrossRef
Tursynbay Y, Zhang J, Li Z, Tokay T, zhumadilov Z, Wu D, Xie Y. Pim-1 kinase as cancer drug target: an update. Biomed Rep, 2016; 4(2):140–6. CrossRef
Wang HL, Cee VJ, Chavez FJ, Lanman BA, Reed AB, Wu B, Guerrero N, Lipford R, Sastri C, Winston J, Andrews KL, Huang X, Lee Mr, Mohr C, Xu Y, Zhou Y, Taskera AS. The discovery of novel 3-(pyrazin-2-yl)-1H-indazoles as potent pan-Pim kinase inhibitors. Bioorg Med Chem Lett, 2015; 25(4):834–40. CrossRef
Watts KS, Dalal P, Murphy RB, Sherman W, Friesner RA, Shelley JC. ConfGen: a conformational search method for efficient generation of bioactive conformers. J Chem Inf Model, 2010; 50(4):534–46. CrossRef
Xu Y, Brenning BG, Kultgen SG, Foulks JM, Clifford A, Lai S, Chan A, Merx S, McCullar MV, Kanner SB, Ho KK. Synthesis and biological evaluation of Pyrazolo[1,5-a]pyrimidine compounds as potent and selective Pim-1 inhibitors. ACS Med Chem Lett, 2014; 6(1):63–7. CrossRef
Zhang Y, Wang Z, Li X, Magnuson NS. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene, 2008; 27:4809–19. CrossRef