
INTRODUCTION
The 17 neglected tropical diseases (NTDs) prioritized by the World Health Organization affect more than 1 billion people worldwide and are endemic in 149 countries (Hotez et al., 2020). Of these NTDs, leishmaniases are a group of diseases caused by the protozoan parasite Leishmania. There are around 20 Leishmania species that are transmitted to humans during the blood meal by infected female phlebotomine sandflies. Leishmaniasis is known to affect over 98 countries, with more than 350 million people at risk. It is estimated that 700,000–1.2 million new cases of leishmaniasis are reported per year. Visceral leishmaniasis (VL), caused by the species Leishmania Donovani, is the most fatal of all infections caused by different leishmanial species which is manifested as a chronic disease in the liver and spleen. It occurs mainly in the Indian subcontinent and East Africa (Tedla et al., 2018). Treatment options for VL are limited. Current treatments mostly rely on drugs that date back to more than 50 years and have certain limitations, such as toxicity (sodium stibogluconate, paromomycin, and miltefosine), increased resistance (pentavalent antimonial chemotherapy), and expensive long-term treatment (Amphotericin B). Thus, the need for new agents that are safe, inexpensive, selective, and effective against resistance is both considerable and urgent. Currently, research on leishmaniasis focuses on the design of specific inhibitors directed toward particular metabolic activities which will damage parasites without affecting the host (Kaur et al., 2010).
For developing specific inhibitors, knowledge of the differences between infective organisms and their respective host’s biochemical pathways, metabolism, and macromolecular structure, as well as detailed characterization of target proteins and macromolecules, is of primary importance (Datta et al., 2008). Trypanosomatid species have been widely studied. Their molecular biology, enzymology, and genome sequencing are well understood. This can help in exploring new targets for combating trypanosomatid infections (Schuttelkopf et al., 2005).
Leishmania species are auxotrophic for folates and pterins. They depend on an exogenous source for folates and biopterins. Inhibition of the enzymes involved in the biochemical cascade can prove to be suitable targets for the treatment of leishmaniasis (Cavazzuti et al., 2008). Dihydrofolate reductase (DHFR) and thymidylate synthase (TS) are the two important enzymes that reduce folates obtained by the organism from an exogenous source, which are utilized by the leishmania species. Along with these enzymes, Pteridine reductase 1 (PTR1), which belongs to the reductase family, catalyzes the Nicotinamide adenine dinucleotide phosphate hydrogen (NADPH)-dependent reduction of folates and pterins. PTR1 reduces oxidized pterins to dihydrobiopterins (DHB) and further to tetrahydrobiopterin and also reduces folates to dihydrofolates and tetrahydrofolates, which are required for the growth of Leishmania. PTR1, thus, acts as a bypass for folate and pterine metabolism when the DHFR-TS system is inhibited. Failure of the DHFR targeting drug therapies against trypanosomatids is due to this enzyme (Chandrasekaran et al., 2016; Kaur et al., 2012). Thus, PTR1 that is responsible for providing essential elements to leishmania for its survival can act as an effective drug target against leishmaniasis.
This study aims at designing PTR1 inhibitors with potential antileishmanial activity. The enzyme PTR1 is a tetramer (subunits A, B, C, and D) (Gourley et al., 2001). Figure 1 shows the 3D structure of the receptor bound to DHB. A series of crystallographic analysis of PTR1 from different leishmania species has been studied. The crystal structure of Leishmania major pteridine reductase 1 (LmPTR1) shows a substrate binding site as a well-defined cleft. In majority of the crystal structures, the pteridine binding sites were identified as π-stacking interactions to Phe 113 and the nicotinamide part of NADPH; an essential hydrogen bond with oxygen atoms of cofactor phosphate group; optional hydrogen bonds to either the hydroxyl group of Ser 111, Tyr 194, or the ribose part of the cofactor (Ferrari et al., 2011; Kaur et al., 2012; Kumar et al., 2008; Mpamhanga et al., 2009). Four pharmacophore features have been identified as key features involved in the inhibitor–PTR1 interaction, which are two H-bond donors, one hydrophobic aromatic feature and one ring aromatic feature. Thus, a structure containing an aromatic ring system, a hydrophobic group, and H-bond acceptors would act as a good substrate for the receptor. These interactions have been made use of to develop potential enzyme inhibitors (Dube et al., 2012; Mpamhanga et al., 2009; Tulloch et al., 2010). Also, heterocyclic compounds, like aminobenzimidazole (Mpamhanga et al., 2009; Spinks et al., 2011), aminobenzothiazole (Mpamhanga et al., 2009), 2,4-diaminopteridine and 2,4-diaminopyrimidine (Tulloch et al., 2010), 1,3,4-thiadiazole (Ferrari et al., 2011), oxadiazoles and triazoles (Cottrell et al., 2004; Ferreira et al., 2007; Rastogi et al., 2006), pyrrolopyrimidine (Khalaf et al., 2014) and pyrimido[1,2-b]pyrimidinone (Kaur et al., 2011), have been studied for PTR1 inhibitory antileishmanial activity.
Blending the data obtained from literature, a series of 2-substituted-5-[(6-substituted-1H-benzimidazol-2yl)methyl] azole derivatives (B1–B14) have been designed which can act as PTR1 inhibitors. Figure 2 shows the general structure of the series designed. Molecular docking studies of 2-substituted-5-[(6-substituted-1H-benzimidazol-2yl)methyl] azole derivatives (B1–B14) were carried out against PTR1. The results of the docking studies gave some valuable information about the interaction of the designed compounds with the receptor. Druglikeness and Absorption, Distribution, Metabolism, Excretion, Toxicity (ADMET) prediction studies were undertaken.
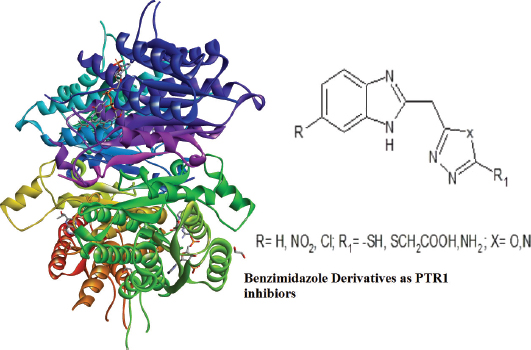 | Figure 1. 3D structure of PTR1 (PDB ID 1E92) complexed with the ligand DHB. [Click here to view] |
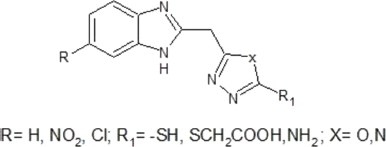 | Figure 2. General structure of 2-substituted-5-[(6-substituted-1H-benzimidazol-2yl)methyl] azole derivatives. [Click here to view] |
MATERIALS AND METHODS
Molecular docking studies
Computational resources
Molecular docking simulations were used to predict the correct binding conformation of the ligand in the enzyme-binding pocket. The scoring function (dock score) comprised steric and electrostatic components of binding parameterized universal force fields. This utility allowed screening a set of compounds for lead optimization. VLifeMDS (Molecular design suite) uses genetic algorithms (GA), piecewise linear pairwise potential function, and grid algorithms to minimize the interaction energy between ligand and receptor.
All molecular modeling studies were carried out using the Molecular Design Suite (VLifeMDS software package, version 4.4; from VLife Sciences, Pune, India), molecular docking carried out by using a dell Personal Computer (PC) with a Pentium IV processor and Windows 7 operating system. Docking studies were carried out using the Ligand docking methology (GRIP) batch docking method implemented in VLifeMDS 4.4 software package.
X-ray crystal structure
The X-ray crystal structure of L. major PTR1 (PDB ID: 1E92) was imported from Protein Data Bank (available at http://www.rcsb.org/). The X-ray crystal structure of the PTR1 domain had a resolution of 2.2â„«.
Protein preparation
The crude Protein Data Bank (PDB) structure of the receptor was refined by completing the incomplete residues. Chloride ions and Adenosine diphosphate (ADP) were deleted. Water molecules were also removed, and hydrogen atoms were added. The optimized receptor was saved as a .mol file and used for docking simulation.
Ligand preparation
The 2D structures of the designed molecules and the reference ligand, methotrexate, were sketched using MarvinSketch 5.11.5 and then converted to 3D structures using the VLifeMDS 4.4 software. The 3D structures were then energy-minimized to an Root mean square (RMS) gradient of 0.01 kcal/mol â„« using Universal Force Field. Conformers of all the designed ligands were selected and the number of seeds used for searching the conformational space was set as 5. All conformers were then energy-minimized to an RMS gradient of 0.01 kcal/mol â„«, and then saved in separate folder.
Docking
Flexible docking algorithm was used which not only predicted the binding mode of a molecule more accurately than rigid body algorithms, but also its binding affinity relative to other compounds (Verkhivker et al., 2000). All conformers were docked using an exhaustive method. The number of placements was fixed to a value of 30 and the rotation angle to a value of 15°. The docking score was used as a scoring function. By rotation angle, the ligand was rotated to obtain different poses. By placements, the method checked for all the 30 possible placements into the active site pocket and picked out few best placements out of 30. For each ligand, all the conformers with their best placements and their docking scores were saved to the output folder. The ligand forming the most stable drug receptor complex was the one which had the minimum docking score (interaction energy) and the scoring interaction energy of the standard drug ligand for comparison. The most stable drug receptor poses were studied for their interactions with the amino acid residues in the active site of the receptor. These interactions involved hydrogen bonding, van der Waal’s interaction, aromatic/π stacking, hydrophobic and other charge interactions.
Druglikeness, bioactivity prediction, and ADMET properties
The in-silico studies helped to determine the activity of the compound when inside the body and served as an important tool for drug discovery and lead optimization.
Molecular descriptors and druglikeliness properties of 2-substituted-5-[(6-substituted-1H-benzimidazol-2yl)methyl] azole derivatives, DHB, and methotrexate (MTX) were analyzed using the Molinspiration tool server (http://www.molinspiration.com, accessed on 21 September 2019), which is based on Lipinski’s rules of five (RO5). The pharmacokinetic properties, such as absorption, distribution, metabolism, excretion, and toxicity, of the compounds were checked using the admetSAR online database (http://lmmd.ecust.edu.cn/admetsar2/, accessed on 05 December 2019) (Cheng et al., 2012). The structures were drawn using Advanced Chemistry Department (ACD)/ChemSketch version 12.0, and Simplified molecular-input line-entry system (SMILES) notation data were generated and fed into these softwares to calculate the parameters.
RESULTS AND DISCUSSION
Designing
The series was designed based on the findings reported in the literature related to the active binding site of PTR1, the structural features for effective interaction with the receptor, as well as various pharmacophores that have been explored in this context. The striking features learnt from the literature were that H-bonding interactions and π-stacking interactions are important and necessary for the binding of inhibitors in the active site of PTR1. There are several hydrophilic and hydrophobic regions in the active site of LmPTR1. The hydrophobic region is at the core of the active site surrounded by hydrophilic regions. Studies on interactions of 1, 3, 4-thiadizol-2-yl-amine derivatives with PTR1 revealed some important points related to the active site. It was learnt that the hydrophobic region had enough space to accommodate various substituted aromatic ring systems. Increasing the hydrophobic quotient by adding a benzene ring to give benzothiazole showed better overlap with the hydrophobic region when compare to the thiadiazole ring system. Scaffolds, such as aminobenzimidazole and aminobenzothiazole, show selective binding to LmPTR1. Taking all these studies as the basis and using the principle of bioisosterism, benzimidazole derivatives bearing a substituted azoles ring fulfilling the hydrophobic, aromatic, and hydrophilic requirements for binding in the active site of PTR1 were designed. Substitutions on the azole ring consisted of functional groups that can act as H-bond acceptors. Moreover, substitutions on the benzimidazole ring were incorporated to study the effect of these substituents on binding. 2-substituted-5-[(6-substituted-1H-benzimidazol-2yl) methyl] azole derivatives (B1–B14) are enlisted in Table 1.
Molecular docking
The elucidation of interactions between PTR1 and the designed series is crucial to check whether the compounds are able to mimic the binding mode of the substrate. Molecular docking was carried out to evaluate the interactions of designed compounds against the L. Major PTR1 crystal structure (PDB Code: 1E92) by using VLifeMDS software package, version 4.4. L. major and L. donovani enzymes share 91% sequence identity and homology modeling and later suggest a structural relationship in and around the active site. Docking studies showed that the designed molecules fit well in the active site pocket made up of the following key residues: Arg 17, Leu 18, Ser 111, Phe 113, Tyr 191, Pro 224, Gly 225, Ser 227, Leu 229, and Val 230. The interactions were compared to the original ligand, DHB, and reference molecule, methotrexate (MTX). As per the available crystallographic data, the substrate DHB binds to LmPTR1 by forming an extended network of H-bonds and aromatic interaction with a co-factor and Phe 113. Similar interactions are observed in MTX with LdPTR1 involving hydrogen bonding interaction of pteridine moiety with Ser 111, Tyr 194, and Arg 17 of LdPTR (Kaur et al., 2011). The designed molecules mimic the key interactions, which include hydrogen bonding, hydrophobic, aromatic, and van der Waal’s interactions. It was observed that the benzimidazole ring, the azole ring and the spacer methylene group were involved in hydrophobic interactions and aromatic π-stacking interaction majorly with amino acid residues Leu 229, Val 230, Phe 113, respectively. The hydrophilic substituents on the azole ring (–SH, −SCH2COOH, −NH2) formed an H-bond with the active site with residues Arg17 and Ser111 in most of the structures, which is similar to the interaction of DHB with the active site. The 2D and 3D interaction images were developed using Discovery studio visualizer v20 (Biswal et al., 2019). Figures 3–6 show the 2D and 3D interactions of the original ligand (DHB), the reference ligand (MTX), compounds B5 and compound B12, respectively, with the active site of PTR1.
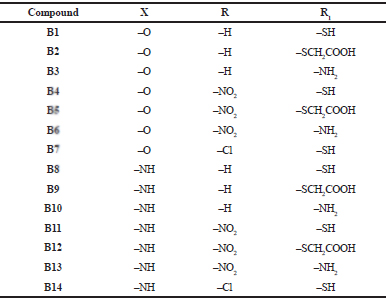 | Table 1. Designed series (B1–B14) with the substituents. [Click here to view] |
The docking scores of the molecules (B1–B14) are presented in Table 2 and their interactions with the amino acids in the active site of PTR1 are listed in Table 3. Compounds B5 and B12 showed the lowest interaction energy of −61.5232 and −62.5897, respectively, which is comparable with the docking scores of the original ligand, DHB, which is −68.4502. It was observed that the length of substituents and H-bond acceptor functionalities on the azole ring played a crucial role in the interaction, which is reflected in the docking scores. Derivatives with a larger group, like SCH2COOH, displayed good affinity when compared to –SH and –NH2. The substituents on the benzimidazole ring (–Cl and –NO2) were more favorable for hydrophobic interactions when compared to the unsubstituted benzimidazole derivatives.
Druglikeness, bioactivity prediction, and ADMET Studies
Absorption, distribution, metabolism, and elimination (ADME) properties play a major role in the success or failure of candidate molecules in drug development. Poor properties can limit the exposure of the compound to the target protein. Toxicity is another very important factor which often overshadows the ADME behavior. Lipinski’s RO5 is useful in assessing the bioavailability of the orally administered compound. According to the rule, a molecule bearing hydrogen bond donors <5 (OH and NH groups), hydrogen bond acceptors <10 (N and O atoms), molecular weight <500, and calculated logP <5 have a great potential for oral bioavailability (Lipinski et al., 2001). Other parameters that give the measure of absorption include water solubility (Log S), topographical polar surface areas (TPSA), human intestinal absorption (HIA), Caco-2 permeability, and blood–brain barrier (BBB) penetration (Ouassaf et al., 2018). For distribution and transport of drugs, generally, only the unbound drug molecule is available for diffusion or transport across cell membranes and for interaction with a pharmacological target. P-glycoprotein (P-gp), an efflux membrane transporter, is widely distributed throughout the body and is responsible for limiting cellular uptake and the distribution of xenobiotic and toxic substances. Many drugs are substrates for this transporter. This transporter can impede the absorption, permeability, and retention of the drugs by extruding them out of the cells (Amin, 2013). When it comes to drug metabolism, most of the drugs are metabolized by the cytochrome P450 class of enzymes. Majority of drug molecules are metabolized by the two isoforms: 2D6 and 3A4. Thus, it is necessary to know whether the designed compounds are substrates for these enzymes. At the same time, the molecules should not inhibit these enzymes as it may hinder the metabolic fate of other drugs. Toxicity profiles of the compounds can be understood by studying the mutagenicity, carcinogenicity, acute oral toxicity, and LD50 in rats (Belal, 2018).
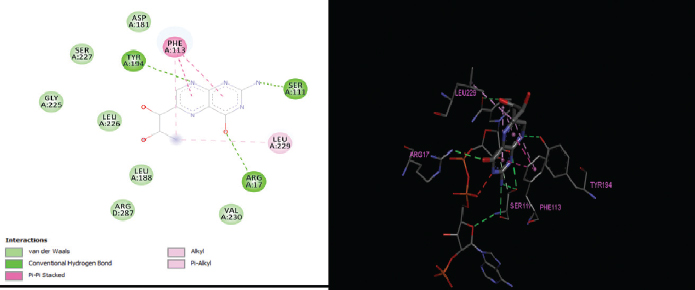 | Figure 3. 2D and 3D interactions of the original ligand DHB with active site residues of PTR1. [Click here to view] |
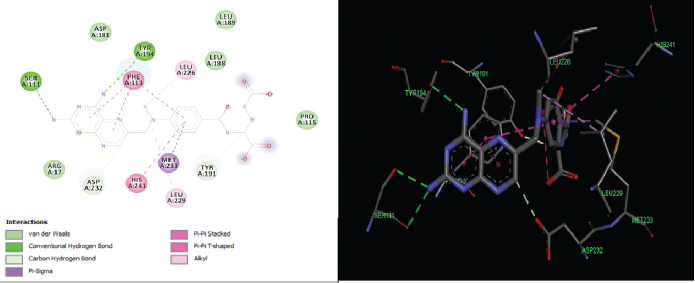 | Figure 4. 2D and 3D interactions of the reference ligand MTX with active site residues of PTR1. [Click here to view] |
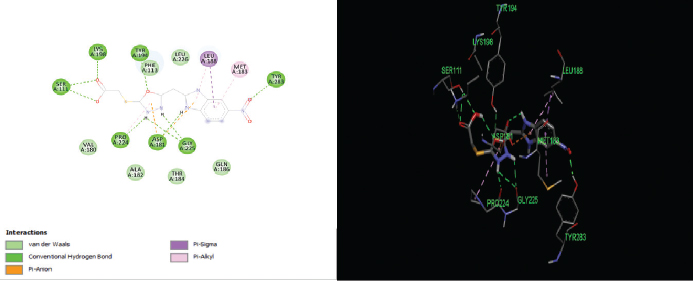 | Figure 5. 2D and 3D interactions of compound B5 with active site residues of PTR1. [Click here to view] |
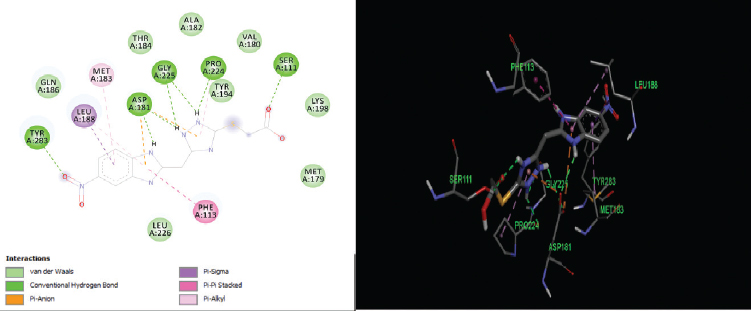 | Figure 6. 2D and 3D interactions of compound B12 with active site residues of PTR1. [Click here to view] |
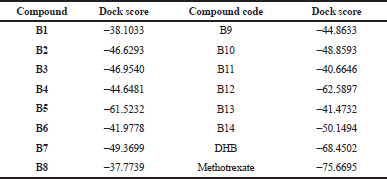 | Table 2. Docking scores of the designed compounds. [Click here to view] |
The designed molecules were checked for the druglikeness (molecular properties) and bioactivity using an online server database, Molinspiration Cheminformatics software. admetSAR, a free online server, was used to predict ADMET properties. All the molecules (B1–B14) showed no violation from Lipinski’s RO5. All compounds followed Veber’s rule as they have rotatable bonds ≤10 and TPSA ≤140° (except for compounds B5 and B12). It indicates that most compounds may have good oral absorption (Veber et al., 2002). While the substrate (DHB) and reference standard MTX show violation from the rules, they both have negative logP values, indicating high affinity to the aqueous phase and high hydrophilicity quotient. Table 4 presents the details of druglikeness studies by using the Molinspiration software. HIA values were found to be 0.9 and above, indicating good intestinal absorption. The Caco-2 cell permeability was found to be moderate, with some molecules having poor permeability values. DHB and MTX were found to have poor Caco-2 cell permeability and may have poor intestinal absorption. All the compounds were found to have high BBB permeability, as well as good oral bioavailability. Log S value indicates water solubility. The lesser the log S value the greater the solubility (Nisha et al., 2016). All compounds displayed log S values in the range of −3.2 to −1.8, indicating good water solubility. Overall, the compounds showed good absorption, distribution, and permeability through biological membranes.
Compounds were found to be non-substrates and non-inhibitors of the P-glycoprotein efflux transporter, and thus will not be extruded out of the cell, and the absorption and permeability of the compounds will not be impeded. Table 5 presents the absorption and distribution profiles of the series. With regard to metabolism, none of the compounds were found to be substrate or inhibitor of CYT2D6 isoform, while for CYT3A4, mixed data was obtained. Some compounds seemed to be both a substrate as well as an inhibitor of this isoform, while some were neither substrate nor inhibitor and some were either substrates or inhibitors. AMES toxicity test is employed to know whether a compound is mutagenic or not. Compounds B1–B3, B8–B10, and B14 displayed negative AMES values, which mean they are non-mutagenic. Most of the compounds were found to be non-carcinogenic, except compounds B4–B6 and B13. Mutagenicity and carcinogenicity can be attributed to the nitro substituents present in the above-mentioned compounds. All compounds showed lower oral acute toxicity than the reference MTX. LD50 is a dose that causes death of 50% of the test population. LD50 of the compounds was found to be relatively higher (ranging from 1.693 to 2.284 mol/kg) and can be considered to be safe. Table 6 presents the metabolism and toxicity data for the series obtained by the admetSAR tool.
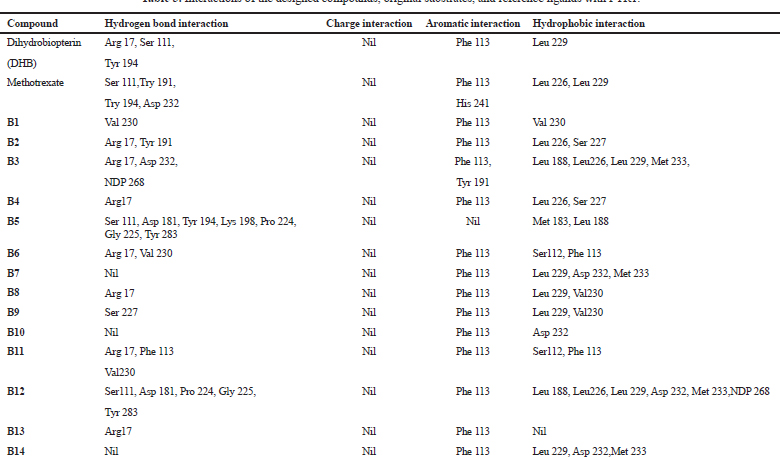 | Table 3. Interactions of the designed compounds, original substrates, and reference ligands with PTR1. [Click here to view] |
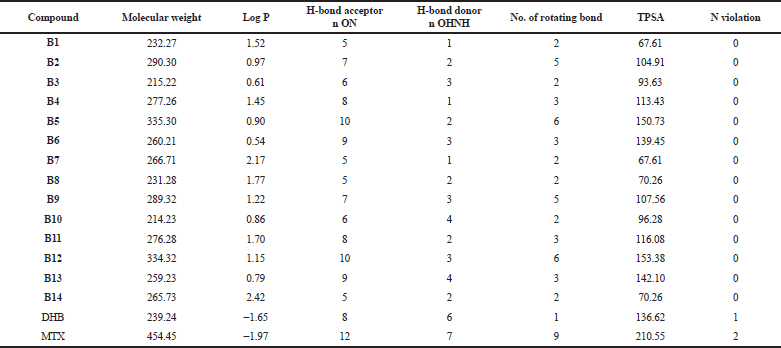 | Table 4. Druglikeness properties of the series using the Molinspiration software. [Click here to view] |
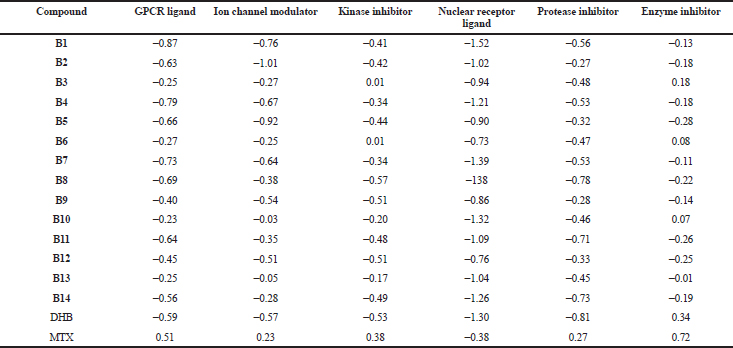
The bioactivity scores of the designed compounds as G-protein coupled receptor (GPCR) ligand, ion channel modulator, nuclear receptor ligand, a kinase inhibitor, protease inhibitor, and enzyme inhibitor were studied and are summarized in Table 7. A molecule having a bioactivity score of more than 0.00 is most likely to exhibit considerable biological activity, while values −0.50–0.00 are expected to be moderately active and if the score is less than −0.50, it is presumed to be inactive (Ertl et al., 2000). Bioactivity scores are more towards 0.0 for enzyme inhibition as compared to other mechanisms. Compounds B3, B6, and B10 exhibited bioactivity scores more than 0.00 for enzyme inhibition, thus they can be considered to exhibit significant biological activity by the above-mentioned mechanism. DHB displayed a bioactivity score of 0.34 under enzyme inhibition mechanism. The remaining compounds gave bioactivity score between −0.28 and −0.01. These scores justify the rationale behind designing the series as PTR1 inhibitor. MTX displayed positive scores in most of the heads, indicating that it exerts physiological action by different mechanisms. From the above set of studies, it was observed that compounds B5 and B12 exhibited the best docking scores, but are associated with potential mutagenicity and carcinogenicity. Compounds B2, B3, and B10 have comparable docking scores and satisfactory druglikeness, bioactivity scores, and ADMET properties. It can be summarized that the substituent on the benzimidazole ring and substitution on the azole ring have an important role in affinity to the receptor, as well the ADMET properties which can be explored in the future.
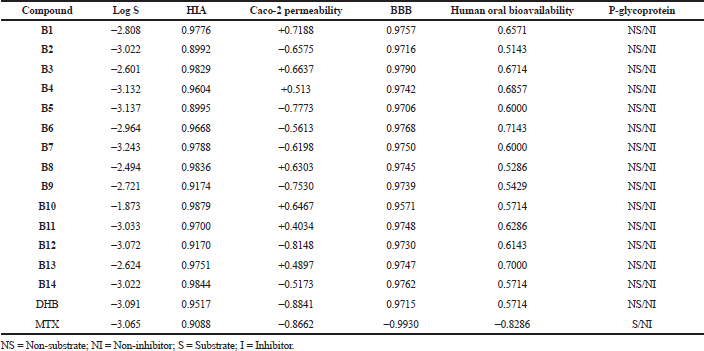 | Table 5. Absorption and distribution profile of the series by using the admetSAR tool. [Click here to view] |
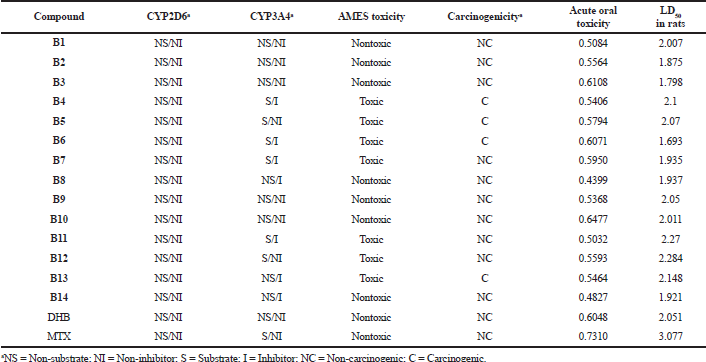 | Table 6. Metabolism and toxicity profile of the series by using the admetSAR tool. [Click here to view] |
 | Table 7. Bioactivity scores of the series by using the Molinspiration software. [Click here to view] |
CONCLUSION
With a view to develop PTR1 inhibitors, a series of 2-substituted-5-[(6-substituted-1H-benzimidazol-2yl)methyl] azole derivatives (B1–B14) was designed. The bioactivity prediction studies substantiated the designed series as enzyme inhibitors. These compounds were subjected to docking studies which showed significant binding of the compounds with PTR1. It was observed that substituents on the benzimidazole ring (−Cl, −NO2) were favorable for binding in the active site of PTR1, as seen in compounds B5 and B12, but may be responsible for mutagenicity and carcinogenicity. Thus, other substituents that are devoid of the toxicity can be explored. Compounds B2, B3, and B10 were found to possess comparable docking scores and satisfactory druglikeness, bioactivity scores, and ADMET properties, and can be considered as good oral drug candidates. In the future, based on the findings of docking studies and in-silico studies, the new series will be designed with structural modifications that would further improve receptor binding and toxicity profiles. The series will be synthesized and tested for antileishmanial activity.
ACKNOWLEDGMENTS
The authors acknowledge VLife Sciences Technologies Pvt. Ltd., Pune, India, for providing the software facility.
AUTHORS’ CONTRIBUTION
Dr. Rakesh Somani, Dr. Devender Pathak, and Mrs. Shraddha Phadke contributed equally to the conception and design, critical review, statistical analysis, administrative and technical support, as well as final approval of the article. Dr. Rakesh Somani and Dr. Devender Pathak also supervised the process of article preparation. Drafting the article, data acquisition, interpretation, communication, and correspondence were conducted by Mrs. Shraddha Phadke.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
FINANCIAL SUPPORT AND SPONSORSHIP
None.
REFERENCES
Amin Md L. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights, 2013; 7:27–34. CrossRef
Belal A. Drug likeness, targets, molecular docking and ADMET studies for some indolizine derivatives. Pharmazie, 2018; 73:635–42.
Biswal AR, Venkatraghavan R, Pazhamalai V, Romauld IS. Molecular docking of various bioactive compounds from essential oil Trachyaspermum ammi against the fungal enzyme Candidapepsin – 1. J Appl Pharm Sci, 2019; 9:021–32. CrossRef
Cavazzuti A, Paglietti G, Hunter WN, Gamarro F, Piras S, Loriga M, Allecca S, Corona P, McLuskey K, Tulloch L, Gibellini F, Ferrari S, Costi MP. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc Natl Acad Sci U S A, 2008; 105:1448–51. CrossRef
Chandrasekaran S, Veronica J, Gundampati RK, Sundar S, Maurya R. Exploring the inhibitory activity of Withaferin-A against Pteridine reductase-1 of L. donovani. J Enzyme Inhib Med Chem, 2016; 31:1029–37. CrossRef
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Tang Y. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model, 2012; 52:3099–105. CrossRef
Cottrell DM, Capers J, Salem MM, DeLuca-Fradley K, Croft SL, Werbovetz KA. Antikinetoplastid activity of 3-aryl-5-thiocyanatomethyl-1, 2, 4 - oxadiazoles. Bioorg Med Chem, 2004; 12:2815–24. CrossRef
Datta AK, Datta R, Sen B. Antiparasitic chemotherapy: tinkering with the purine salvage pathway. Adv Exp Med Biol, 2008; 625:116–32. CrossRef
Dube D, Periwal V, Kumar M, Sharma S, Singh TP, Kaur P. 3D-QSAR based pharmacophore modeling and virtual screening for identification of novel pteridine reductase inhibitors. J Mol Model, 2012; 18:1701–11. CrossRef
Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem, 2000; 43:3714–7. CrossRef
Ferrari S, Morandi F, Motiejunas D, Nerini E, Henrich S, Luciani R, Venturelli A, Lazzari S, Calò S, Gupta S, Hannaert V, Michels PAM, Wade RC, Costi MP. Virtual screening identification of nonfolate compounds, including a CNS drug, as antiparasitic agents inhibiting pteridine reductase. J Med Chem, 2011; 54:211–21. CrossRef
Ferreira SB, Costa MS, Boechat N, Bezerra RJS, Genestra MS, Canto-Cavalheiro MM, Kover WB, Ferreira VB. Synthesis and evaluation of new difluoromethyl azoles as antileishmanial agents. Eur J Med Chem, 2007; 42:1388–95. CrossRef
Gourley DG, Schuttelkopf AW, Leonard GA, Luba J, Hardy LW, Beverley SM, Hunter WN. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasite. Nat Struct Biol, 2001; 8:521–5. CrossRef
Hotez PJ, Aksoy S, Brindley PJ, Kamhawi S. World neglected tropical diseases day. PLoS Negl Trop Dis, 2020; 14:e0007999. CrossRef
Kaur J, Dube D, Ramchandran R, Singh P, Singh N. Thianthrene is a novel inhibitor of Leishmania donovani pteridine reductase 1 (PTR 1). J Mol Biol, 2012; 1:68–75.
Kaur J, Sundar S, Singh N. Molecular docking, structure–activity relationship and biological evaluation of the anticancer drug monastrol as a pteridine reductase inhibitor in a clinical isolate of Leishmania donovani. J Antimicrob Chemother, 2010; 65:1742–8. CrossRef
Kaur J, Kumar P, Tyagi S, Pathak R, Batra S, Singh P , Singh N. In silico screening, structure actvityy relationship, and biological evaluation of selective pterdine reductase inhibitors targeting visceral leishmaniasis. Antimicrob Agents Chemother, 2011; 55:659–66. CrossRef
Khalaf AI, Huggan JK, Suckling CJ, Gibson CL, Stewart K, Giordani F, Barrett MP, Wong PE, Barrack KL, Hunter WN. Structure-based design and synthesis of antiparasitic pyrrolopyrimidines targeting pteridine reductase 1. J Med Chem, 2014; 57:6479–94. CrossRef
Kumar P, Kumar A, Verma SS, Dwivedi N, Singh N, Siddiqi MI, Tripathi R, Dube A, Singh N. Leishmania donovani pteridine reductase 1: biochemical properties and structure-modeling studies. Exp Parasitol, 2008; 120:73–9. CrossRef
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development Settings. Adv Drug Deliv Rev, 2001; 46:3–26. CrossRef
Mpamhanga CP, Spinks D, Tulloch LB, Shanks EJ, Robinson DA, Collie IT, Fairlamb AH, Wyatt PG, Frearson JA, Hunter WN, Gilbert IH, Brenk R. One scaffold, three binding modes: novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J Med Chem, 2009; 52:4454–65. CrossRef
Nisha CM, Kumar A, Nair P, Gupta N, Silakari C, Tripathi T, Kumar A. Molecular docking and in-silico ADMET study reveals acylguanidine 7a as a potential inhibitor of βSecretase. Adv Bioinformatics, 2016; 2016:1–6. CrossRef
Ouassaf M, Belaidi S, Lotfy K, Daoud I, Belaidi H. Molecular docking studies and ADMET properties of new 1.2.3 triazole derivatives for anti-breast cancer activity. J Bionanosci, 2018; 12:1–11. CrossRef
Rastogi, N, Singh VR. Microwave mediated aminomethylation and antileishmanial activity of 2-{4'-(2",4"-Dichlorobenzyloxy)-phenyl}-1,3,4-oxadiazolin-5-thiones and 3-{4'-(2",4"-Dichlorobenzyloxy)-phenyl}-4-phenyl-1,2,4-triazolin-5- thiones. Indian J Heterocycl Chem, 2006; 16:5–8.
Schuttelkopf AW, Hardy LW, Beverley SM, Hunter WN. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J Mol Biol, 2005; 352:105–16. CrossRef
Spinks D, Ong HB, Mpamhanga CP, Shanks EJ, Robinson DA, Collie IT, Read KD, Frearson JA, Wyatt PG, Brenk R, Fairlamb AH, Gilbert IH. Design, synthesis and biological evaluation of novel inhibitors of trypanosoma brucei pteridine reductase 1. Chem Med Chem, 2011; 6:302–8. CrossRef
Tedla DW, Bariagabr FH, Abreha HH. Incidence and trends of leishmaniasis and its risk factors in humera, Western Tigray. J Parasitol Res, 2018; 2018:8463097. CrossRef
Tulloch LB, Martini VP, Iulek J, Huggan JK, Lee JH, Gibson CL, Smith TK, Suckling CJ, Hunter WN. Structure–based design of pteridine reductase inhibitors targetting African sleeping sickness and leishmaniasis. J Med Chem, 2010; 53:221–9. CrossRef
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem, 2002; 45:2615–23. CrossRef
Verkhivker GM, Bouzida D, Gehlhaar DK, Rejeto PA, Arthurs S, Colson AB, Freer ST, Larson V, Luty BA, Marrone T, Rose PW. Deciphering common failures in molecular docking of ligand-protein complexes. J Comput Aided Mol Des, 2000; 14:731–51 CrossRef