INTRODUCTION
Diabetes is a group of metabolic disease, in which a person has high blood sugar, either because the pancreas does not produce enough insulin or cells do not respond to the insulin that is produced (David & Dolores, 2011). The WHO has predicted that the major burden will occur in developing countries. The studies conducted in India in the past decade have highlighted that not only there is a high prevalence of diabetes but also it is increasing rapidly in the urban population. It is estimated that there are approximately 33 million adults with diabetes in India. This number is likely to increase to 57.2 million by the year 2025 (Manisha et al., 2007).
Beta-cell destruction in type I diabetes mellitus (DM) is prominent that leads to insulin deficiency. Glucose metabolism is affected in the body and accumulates and gives rise to multiple complications. In patients with DM, years of poorly controlled hyperglycemia lead to multiple, primarily vascular complications that affect small vessels (microvascular), large vessels (macrovascular), or both. Pancreatic islets are thought to play a key role in the pathophysiology of diabetes through the failure of islet beta cells to secret the sufficient quantities of insulin to regulate blood glucose and are, therefore, a key focus of diabetes research (Donath & Halban, 2004).
The use of natural products in modern medicine even though widespread in curing or preventing diseases lacks scientific evidence in most of the cases as to whether it is to be used as a plant or its active constituents (Bhonde et al., 1999; Dittrich & Dorsche, 1978; Singh et al., 2000). Several drugs have been discovered and are in use, which either increase insulin secretion or increase the utilization of glucose by the peripheral tissues. However, to date, no drug has been discovered, which can regenerate or protect the islets. Alternative regenerative options such as the use of stem cells are there, but their clinical application is not validated.
Momordica dioica has also the same potent as the other natural drugs as already reported in various scientific papers; the antidiabetic activity of fraction of saponin isolated from the fruits of M. dioica showed a reduced glucose level in alloxan-induced diabetic rats and enhanced insulin sensitivity (Firdous et al., 2009). Steroidal saponin showed an improvization in lipid profile, which lowers the levels of HbA1c and increases serum insulin, reversible of beta-cell degeneration in vivo (Jha et al., 2019). Antidiabetic and insulin secretagogues activity of M. dioica has been reported earlier, but no study has been done so far on islet protection in vitro. There may not be any better drug if one can stimulate the regeneration of islets containing insulin-producing cells. This can put the diabetics of the antidiabetic drugs. The isolated phytoconstituents of the plants if found promising in ameliorating the severity of diabetes can be further exploited for the betterment of mankind.
This study was focused on the optimization of the most active method for the isolation of islets from rat pancreas and explored the influence of the saponin of M. dioica (SMD) on the isolated islets in various simulated diabetic conditions (Fig. 1).
MATERIALS AND METHODS
Saponin isolation and characterization
The extraction and isolation of saponin from the fruits of M. dioica were already done and reported in the previous paper (Jha et al., 2019). The pure saponin was further studied and characterized by spectroscopic techniques such as high-performance liquid chromatography (HPLC), liquid chromatography-mass spectroscopy (LCMS), Fourier transmission infrared (FT-IR), and nuclear magnetic resonance (1H-NMR) spectroscopy.
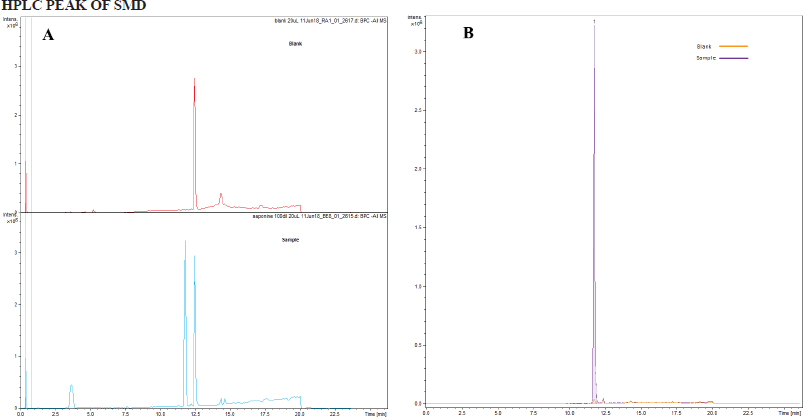
HPLC of SMD
The sample was diluted 100 times with 5% acetonitrile (ACN) in water. Dionex 3,000 Ultimate was used as the Liquid chromatography (LC). Buffer A was 0.1% formic acid in water and buffer B was 0.1% formic acid in ACN. The flow rate was 500 μl/minutes. The gradient was as follows: 0–10 minutes increase from 5% B to 98% B and 10–20 minutes hold at 98% B and finally come back to 5% B. A Hypersil GOLD C18 (Thermo Fisher Scientific, Waltham, MA) column was used for the separation (150 mm × 4.6 mm, particle size of 3 μm). The column was kept at 35°C. UV detection was carried out at 280 nm.
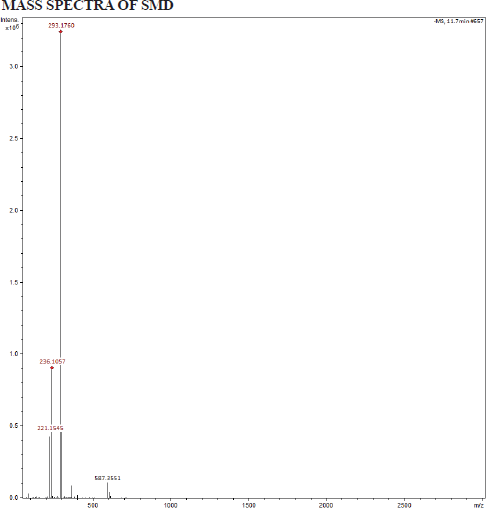
LCMS of SMD
Bruker Impact HD QTOF Mass Spectrometer was used in this LCMS experiment equipped with an electrospray ionization source. [Source: Software and data analysis: Bruker otofControl software (version 3.3 build 18) was used to operate the mass spectrometer].
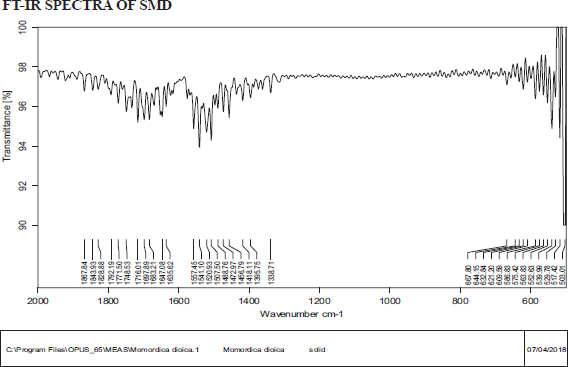
FTIR of SMD
Source: Bruker alpha series was used to operate the FTIR, and the instrument is available in the Analytical Laboratory, Karnataka College of Pharmacy, Bangalore.
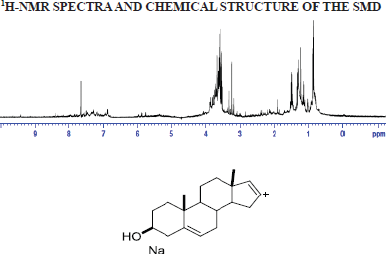
1H-NMR of SMD
NMR spectra were recorded on a Bruker-AV-400 NMR spectrometer at room temperature in MeOD and dimethyl sulfoxide (DMSO), respectively, with Tetramethylsilane (TMS) acting as an internal standard. Chemical shifts (δ) were expressed in parts per million (ppm) with coupling constants (J) in Hertz (Hz).
Animals
Wistar male rats weighing 120–150 g were used for the experiment. The entire study was conducted in accordance with the Committee for the purpose of control and supervision of experiments on animals (CPCSEA) guidelines. All the experiments conducted on the animals were in accordance with the standards set for the use of the laboratory animal use, and the experimental protocols were duly approved by the Institutional animal ethics committee (IAEC) of Karnataka College of Pharmacy, Bangalore (Ref. no: KCP-IAEC2/17-18/15-04-07).
 | Figure 1. Courtesy: Bioprotocol 2016; 6(12) isolation and culture of the islets of Langerhans from rat pancreas. [Click here to view] |
Optimization of islet isolation and culture
Following are methods employed for the optimization of the isolation of rat pancreatic islets:
(A) Pancreas mincing and collagenase type IV digestion
(B) Pancreatic perfusion of collagenase type XI via the common bile duct (CBD) postduodenal occlusion
(C) Pancreas mincing and collagenase type XI digestion followed by cell straining and Ficoll gradient centrifugation.
(D) Pancreas mincing and collagenase type XI digestion followed by cell straining (500 μm), Ficoll gradient centrifugation, and cell straining (70 μm) (Graham et al., 2016; Samaddar et al., 2019).
Pancreas mincing and collagenase type IV digestion
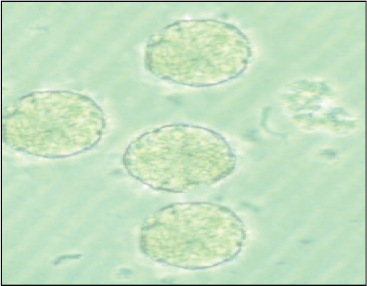
Rat was sacrificed with the high dose of pentobarbital sodium I.P. The pancreas was isolated and then transferred to a sterile polypropylene Petri plate containing sterile Hank’s balanced salt solution (HBSS) with 20 mM of HEPES (N-2-hydroxyethylpiperazine-N-ethanesulfonic acid), 2 mM of CaCl2.2H2O, and penicillin–streptomycin–amphotericin B (100 IU/ml–100 μg/ml–2.5 μg/ml, respectively) solution. The superficial fatty tissues and blood clots were removed by HBSS wash and mincing of the pancreas followed by HBSS wash for three times. The minced tissue mass was transferred to a 50 ml of sterile conical-bottom centrifuge tube containing 5 ml of collagenase type IV (1 mg/ml; HiMedia, Mumbai) in Roswell Park Memorial Institute-1640 (RPMI 1640). The tube was incubated at 37°C in a water bath for 20 minutes with occasional shaking. After 20 minutes, the digestion was stopped by thrusting the tube in ice and adding 10% fetal bovine serum (FBS). The tube was centrifuged at 1,000 rpm for 10 minutes. The cell pellet was resuspended in warm RPMI with 10% FBS and seeded in a T25 culture flask (Nunc, Denmark). The islets were incubated at 37°C in 5% CO2 in a CO2 incubator (Forma, Thermo Scientific, Waltham, MA). Healthy islets (with smooth borders and no dark center) are handpicked using a 200-μl micropipette under the microscope and placed into a Petri plate containing fresh RPMI 1640 medium after 48 hours. These islets were counted and further evaluated for their functionality and viability.
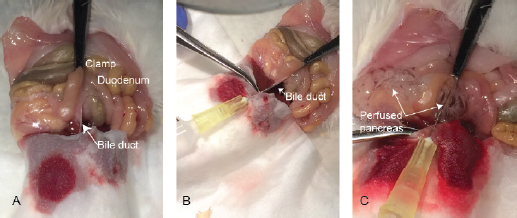
Pancreatic perfusion of collagenase type XI via the CBD post duodenal occlusion
Rat was sacrificed with the high dose of pentobarbital sodium I.P., and an incision was made around the upper abdomen to expose the peritoneum. The liver was flipped over onto a tissue paper, and it was folded over to cover it. Using a curved forceps, the duodenum was located. The confluence, where the CBD enters the duodenum, was located and clamped with a hemostatic forceps to prevent the emptying of collagenase solution in the duodenum. Gently, 3 ml of ice-cold collagenase type XI (Sigma, St. Louis, MO) solution (1 mg/ml) in HBSS was injected through the CBD into the pancreas. The inflated pancreas was gently dissected and placed in a 50-ml conical centrifuge tube containing 2 ml of collagenase type XI solution. The tube was placed in a water bath at 37°C for 15 minutes and briefly shaken 2–3 times by hand during the incubation. After digestion, the tube was removed from the water bath and plunged immediately into ice to stop collagenase digestion, and 15 ml of fresh ice-cold HBSS was added to stop the digestion process completely. The HBSS was removed by centrifuging the tube at 1,000 rpm for 10 minutes, and then, the cell pellet was resuspended in warm RPMI with 10% FBS and seeded in T25 culture flask (Nunc, Denmark). The islets were incubated at 37°C in 5% CO2 in a CO2 incubator (Forma, Thermo Scientific, Waltham, MA). Healthy islets (with smooth borders and no dark center) are handpicked using a 200-μl micropipette under the microscope and placed into a Petri plate containing fresh RPMI 1640 medium after 48 hours. These islets were counted and further evaluated for their functionality and viability.
Pancreas mincing and collagenase type XI digestion followed by cell straining and Ficoll gradient centrifugation
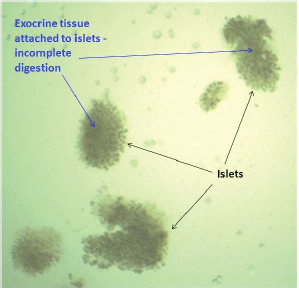
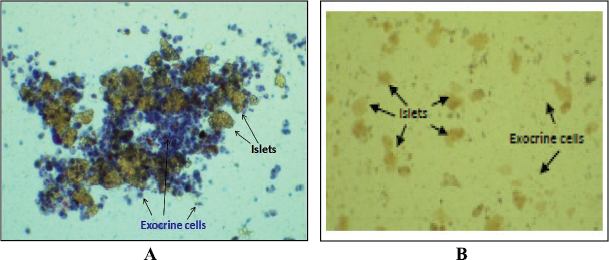
Pancreatic digestion was carried similar to the previous method in a water bath with a slight modification in the digestion medium. Bovine serum albumin fraction V (Sigma, St. Louis, MO) was added to a final concentration of 2%. A 100 μl aliquot of the digestion mixture was withdrawn at a regular interval (5 minutes) and observed under the microscope to monitor the digestion process (Jacqueline et al., 2009). The complete detachment of exocrine tissues from the islets when observed indicates the completion of the digestion process. At this point, the digestion is stopped, and the mixture is centrifuged at 1,000 rpm for 10 minutes. The supernatant is removed, and the cell mass is resuspended in 10-ml warm RPMI. The digested pancreas is filtered through a sterile stainless steel mesh (500-μm pore size). The mesh is washed with 5–10 ml of additional cold RPMI to wash down any residual digested tissue leaving behind the undigested tissue. The tube is centrifuged to obtain the digested pancreas after removing the supernatant and resuspended gently in 10 ml of Histopaque® 1077 (Sigma, St. Louis, MO) at room temperature. This was then overlaid with 5 ml of RPMI 1640 at room temperature extremely gently forming a clear and sharp interface between the two liquids. The tube was centrifuged at 850 × g for 20 minutes with brakes off. It is just beneath the interface where the islets gather after centrifugation. The islets were gently removed from the gradient and transferred to a tube containing RPMI. The medium was removed by centrifugation to wash off the histopaque, and a fresh, warm complete RPMI 1640 containing 10% FBS was added to the islets. The islets were resuspended, seeded into a Petri plate, and incubated at 37°C in 5% CO2 for 48 hours followed by handpicking.
Pancreas mincing and collagenase type XI digestion followed by cell straining (500 μm), Ficoll gradient centrifugation, and cell straining (70 μm)
After density gradient centrifugation (in the previous step), the islets obtained were passed through a prewetted, inverted polypropylene 70-μm cell strainer (Cat. No. CLS431751; Corning, Corning, NY). The strainer was rewashed with fresh medium and turned upside down over a new petri dish containing 15 ml of fresh RPMI to rinse off the captured islets. This method eliminates the exocrine cells leaving behind islets only. The islets were incubated at 37°C in 5% CO2 for 48 hours and handpicked postincubation.
Assessment of islet viability and specificity
The islet viability was assessed by the trypan blue dye exclusion test, and the specificity of islets was determined by dithizone (DTZ) staining (Sigma, St. Louis, MO) (Samaddar et al., 2019). In the trypan blue dye exclusion assay, islets were exposed to the membrane-impermeant dye, trypan blue (0.1% w/v) for 15 minutes at 37°C. Dead and membrane-compromised cells took up the dye and appeared blue while healthy viable cells with intact membrane appeared colorless. For specificity, DTZ stock solution (39 mmol/l) was prepared by dissolving 100 mg of DTZ in 10 ml of DMSO, filtered, aliquoted, and stored at −20°C. Routine staining was carried out by adding 10 μl of DTZ stock to islets suspended in 1 ml of Krebs–Ringer bicarbonate (KRB) buffer (pH 7.4) with (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) (10 mmol/l) and incubated at 37°C for 30 minutes.
Evaluation of islet protective potential assay
Glucose-stimulated insulin secretion assay (GSIS)
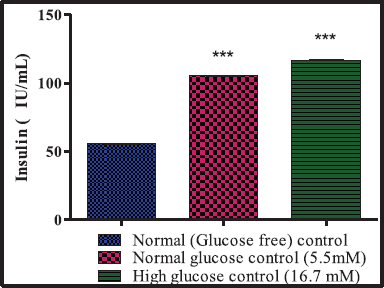
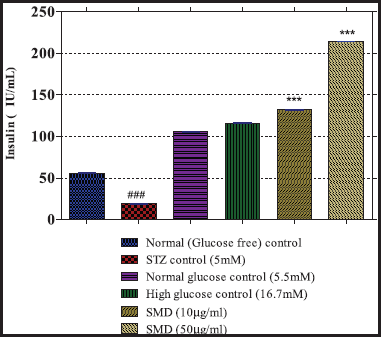
The isolated islets were cultured at 37°C in a humidified atmosphere of 5% CO2 in air in RPMI 1640 medium containing 11.1 mM of glucose, 10% of FBS, and antibiotics (50,000 IU/I penicillin and streptomycin) (Samaddar et al., 2019). The islets were seeded at a concentration of 50 islets per well in 12-well plates (Corning, Corning, NY), and the islets were washed thrice with KRB buffer and preincubated for 1 hour at 37°C. Grouping cells--- The islets were incubated for 1 hour at 37°C, and the aliquots of 10 μl were withdrawn from each well and assayed for insulin.
A total of 50 islets in triplicates (n = 3) were treated in the following manner:
Group 1: Glucose-free control – islets maintained in glucose-free KRB
Group 2: Normal glucose control – islets in 5.5 mM added glucose in KRB
Group 3: High glucose control – islets in 16.7 mM added glucose in KRB
Membrane integrity
After the islets were handpicked and incubated for 24 hours, they were exposed to the membrane-impermeant dye, trypan blue (0·1% w/v) for 15 minutes at 37°C. The presence of dye within cells was determined by light microscopy. Dead cells will take up the dye and appear blue owing to membrane damage, whereas the viable cells will remain unstained.
Effect of SMD on insulin secretion from cultured islets
The islets were cultured and treated with 5 mM of streptozotocin (Sigma, St. Louis, MO) in PBS, pH 7.4 with or without the SMD in low and high dose. Normal and high glucose-treated islets were also maintained in KRB and assayed for insulin.
A group of 50 islets each in triplicate (n = 3) were treated in the following manner:
Group 1: Normal control – islets maintained in glucose-free KRB, pH7.5
Group 2: Streptozocin (STZ) control – islets maintained in glucose-free KRB treated with 5 mM of STZ
Group 3: Normal glucose control – islets in 5.5 mM added glucose in KRB
Group 4: High glucose control – islets in 16.7 mM added glucose in KRB
Group 5: Saponin low dose – islets treated with SMD (10 μg/ml)
Group 6: Saponin high dose – islets treated with SMD (50 μg/ml)
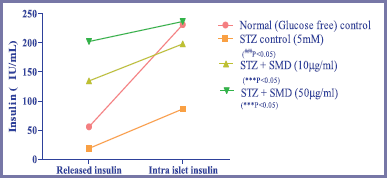
Measurement of released and intra-islet insulin after treatment with STZ and SMD
A group of 50 islets each in triplicate (n = 3) were treated in the following manner:
Group 1: Normal control – islets maintained in glucose-free KRB, pH7.5
Group 2: STZ control – islets maintained in glucose-free KRB treated with 5 mM of STZ
Group 3: Saponin low dose – STZ control + SMD (10 μg/ml)
Group 4: Saponin high dose – STZ control + SMD (50 μg/ml)
Insulin was assayed by sandwich ELISA using a commercially available kit (Cat. No. 10-1250-01, Mercodia, Sweden).
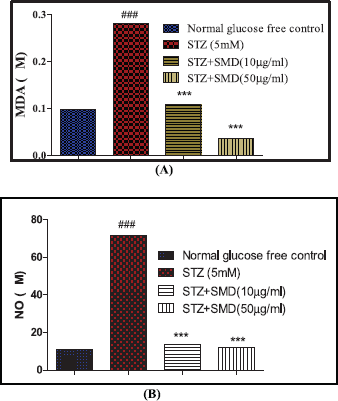
Effect of the SMD on STZ-induced lipid peroxidation and nitric oxide (NO) formation
The assay mixture contained 0.5 ml of cell lysate, 1 ml of 0.5M KCl in 10 mM Tris-HCl, 0.5 ml of 30% trichloroacetic acid, and 0.5 ml of 52 mM thiobarbituric acid (TBA). The assay mixture was heated to 80°C for 30 minutes and after cooling to 0°C centrifuged at 800 x g for 10 minutes. The absorbance of the supernatant was measured at 532 nm. The levels of N-nitrosomethylaniline (NMA) were calculated using the following formula: (Absorbance at 532) – (Absorbance at 600) is Absorbance due to MDA-TBA abduct. The extinction coefficient of this MDA-TBA abduct at 532 nm is 155 mM−1cm−1. The concentration of Malondialdehyde (MDA) (mM) = (A532 – A600)/155
NO produced during STZ and saponin treatment was estimated spectrophotometrically as a formed nitrite (NO2). To measure the nitrite content, 100 μl of the cell lysate was incubated with 100 μl of Griess reagent (1% sulfanilamide in 0.1 mol/l HCl and 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride) at room temperature for 10 minutes. Then, the absorbance was measured at 540 nm using a microplate reader. The nitrite content was calculated based on a standard curve constructed with NaNO2 (Ignarro et al., 1987).
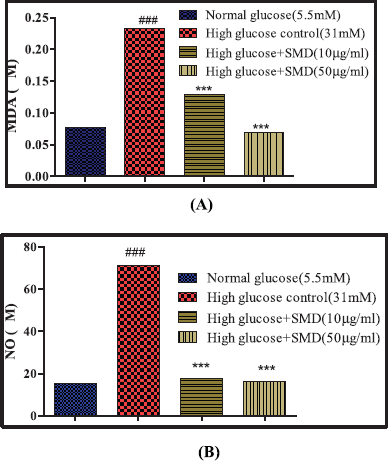
Effect of SMD on high glucose-induced oxidative stress on islets
A group of 50 islets each in triplicate (n = 3) were treated in the following manner:
Group 1: Normal control – islets maintained in normal glucose (5.5 mM) medium
Group 2: High glucose control – islets maintained in high glucose (31 mM) medium
Group 3: Saponin low dose – STZ control + SMD (10 μg/ml)
Group 4: Saponin high dose – STZ control + SMD (50 μg/ml)
The islets were incubated for 24 hours at 37°C in 5% CO2. After incubation, the islets were lysed, as previous, and the lysate was used to estimate the formation of NO and MDA (lipid peroxidation).
Statistical Analysis
The results were expressed as mean ± SEM, and all the statistical comparisons were made by means of the one-way analysis of variance test followed by Tukey's multiple comparison tests (GraphPad Prism v5.0). The p-value < 0.05 was considered to indicate a statistically significant difference.