
INTRODUCTION
Methocarbamol, United States Pharmacopoeia (USP), is a central nervous system (CNS) depressant used in the treatment of skeletal muscle spasms. The exact mechanism to produce skeletal muscle relaxation is not described; however, it is believed to act centrally, probably by general CNS depressant properties (Sweetman, 2011). It is also used as an adjunct in the management of acute painful musculoskeletal conditions during physical therapy (El-Yazbi et al., 2019). Its mechanism of action is also comparable to that of carbamates, through the inhibition of the synaptic acetylcholinesterase at the neuromuscular junction. Methocarbamol does not have a direct impact on the contractile mechanism of striated muscle unlike, carbamates. The onset of action of methocarbamol is approximately 30 minutes because of its rapid intestinal absorption after oral administration and eliminated in the urine in low concentrations.
Methocarbamol is a white to off-white powder, sparingly soluble in chloroform and water, soluble in propylene glycol, and alcohol upon heating and insoluble in n-hexane and benzene. Under the trade name Robaxin®, methocarbamol 500 mg is a light orange, round, film-coated tablet containing excipients, such as FD&C Yellow 6, corn starch, hypromellose, magnesium stearate, hydroxyl propyl cellulose, povidone, propylene glycol, polysorbate 20, sodium lauryl sulfate, saccharin sodium, stearic acid, sodium starch glycolate, and titanium dioxide.
Methocarbamol belongs to the anisoles class of organic compounds, either containing or derivatives of methoxybenzene. The IUPAC name of methocarbamol is [2-hydroxy-3-(2-methoxyphenoxy) propyl] carbamate with a molecular formula of C11H15NO5 (Fig. 1a) and molecular weight of 241.24 g/mol. Methocarbamol is a structural analog of guaifenesin and was derived from guaifenesin by carbamoylation reaction (O’Neil, 2006). During the amination of guaifenesin using phosgene in ethanol, β-isomer of methocarbamol (Fig. 1b) is produced as a potential process-related impurity and was observed consistently in high-performance liquid chromatography (HPLC) analysis, along with the final end product and starting material (Farsam et al., 1999; Raju et al., 2013; Yale et al., 1950) (Fig. 2).
 | Figure 1. Chemical structures and IUPAC nomenclature of (a) methocarbamol and (b) its β-isomer. [Click here to view] |
The purity of both active pharmaceutical ingredients (APIs) and formulations is one of the most challenging tasks for chemists in the chemical and pharmaceutical industry. The purity depends on various factors, such as raw materials, methods of manufacture, purification process, and storage. The presence of impurities in both APIs and finished formulations could degrade its therapeutic efficacy, cause unwanted side effects, and affects its stability. Various pharmacopoeias have published maximum allowed limits for drug-related impurities in both APIs and formulated APIs (Desai et al., 2011; Raju et al., 2013). The permissible limits of methocarbamol and isomer related impurities are official in the USP (The United States Pharmacopeia, 1984). Different analytical methods are published to determine the methocarbamol alone (Walash et al., 2011, 2012; Zha and Zhu, 2010) or in combination with other drugs (Elkady, 2010; Erk et al., 2001) or its enantiomers (Alessi-Severini et al., 1992; Farsam et al., 1999). There is no single analytical method so far reported determining the β-isomeric related impurities of methocarbamol.
This is the first report to our knowledge to discuss the formation, identification, isolation, and characterization of β-isomer of methocarbamol as a potential process-related impurity. As per the earlier literature, methocarbamol is either determined in their binary mixtures or their mixtures with other drugs. The later has been determined by ratio spectra, reverse phase HPLC (RP-HPLC), and second derivative spectrophotometric methods (Ali et al., 2012a, 2012b; Erk et al., 2001; Walash et al., 2012). Hence, the present work was aimed to isolate and purify the impurity and to characterize it by nuclear magnetic resonance (NMR) and mass spectrometry. The HPLC investigation of the methocarbamol has also been performed. The US Food and Drug Administration defines the stringent acceptance criterion for the quality of APIs, intermediates, and impurities. The possible process-related and degradation impurities are crucial as per regulatory requirement, for the safety assessment during a manufacturing process. It is compulsory to find and characterize the impurities in the pharmaceutical products if existing above the accepted limit of 0.1%. This investigation deals with separation and purification followed by structure elucidation of the impurity during the synthesis of methocarbamol from guaifenesin.
 | Figure 2. Synthesis of methocarbamol from guaifenesin. (a) methocarbamol and (b) β-isomer of methocarbamol. [Click here to view] |
MATERIALS AND METHODS
Reagents and samples
HPLC-grade acetonitrile (ACN) and methanol were obtained from Merck (E. Merck, Mumbai, India). Methocarbamol (99.5% pure) and β-isomer were supplied by Shasun Chemicals and Drugs Ltd., Chennai, India. Deuterated Dimethyl Sulfoxide (DMSO-d6), deuterated chloroform (CDCL3), tetramethylsilane (TMS) and all other reagents, chemicals were purchased from Sigma-Aldrich (St. Louis, USA). The water used was collected from a Milli-Q purification system (Millipore Corp., Bedford, USA). The non-aqueous solvent operations were conducted in dry glassware under the inert nitrogen ambience.
Synthesis of methocarbamol and its β-isomer
The initial compound, 122 g of guaifenesin was dissolved in 450 ml of isopropanol and ammonia gas maintaining the reaction at 20°C–25°C for 4–8 hours which lead to the generation of the compound a (methocarbamol), and compound b (β-isomer of methocarbamol) simultaneously. Compound a as a solid, three times by weight was isolated and crystallized from ethanol to give 110 g pure compound a. (Fig. 2a and b).
Mass spectrometry
Liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) analysis has been examined by Waters Micromass Mass Spectrometer using a turbo ion spray interface (negative ionization mode). The declustering potential at 70 V, ion source voltage at 5,500 V, entrance potential at 10 V, and focusing potential at 400 V was set as mass spectral parameter with air as a nebulizer gas under pressure of 40 pounds per square inch (psi) and nitrogen as a curtain gas at a pressure of 25 psi. For LC-MS identification of the impurity, Inertsil ODS-3V (250 mm length with 4.6 mm in diameter with, 5 μm particle size, GL Sciences Inc., Tokyo, Japan) column was used for the separating and isolating the impurity and parent compound with the acceptable resolution between the two chromatographic peaks. Mobile phase composed with 0.1% (v/v) formic acid in water as solvent-A and ACN as solvent-B in the proportion of 95:5 (v/v), respectively. The rate of flow was set at 1.2 ml/minutes with splitting.
NMR spectroscopy
All 1H NMR spectra were generated using a 300 MHz NMR Spectrometer (Bruker NMR Instruments, Oxford, UK). CDCl3 and DMSO-d6 solvents were used and TMS was used as an internal standard.
FT-IR spectroscopy and melting range
FT-IR spectra of neat samples (without solvent) was measured using a attenuated total reflectance FT-IR spectrophotometer (Spectrometer IR Prestige-21, Shimadzu Corporation, Japan) in the spectral range of 600–4,000 cm−1 with 20 scans and 2 cm−1 resolution. Melting point ranges was determined using a melting point apparatus (Electrothermal 9100, Thermo Fisher Scientific, UK).
Analytical HPLC conditions
The analysis was conducted on an Agilent HPLC system with the Photodiode array (PDA) detector. The instrument equipped with a quaternary gradient pump along with an auto- sampler was used for the sample analysis. The chromatographic data were processed by using Chemstation software. The separation of methocarbamol with β-isomer was achieved on Inertsil ODS-3V (250 × 4.6 mm, 5 μ) column with high resolution. The eluents were with the flow rate of mobile phase 1.0 ml/minute were monitored at 274 nm. The RP-HPLC method in isocratic mode of elution was optimized to produce the desired separation of impurity from methocarbamol APIs. The mixture of 6.8 g of KH2PO4 in water adjusted the pH 4.5 with H3PO4 as solvent-A and methanol were chosen as the solvent in the ratio of 75: 25 (v/v), respectively, as the mobile phase. Chromatography was performed under ambient conditions using the flow rate of 1.0 ml/minutes with an injection volume of 20 μl. The chromatographic run time was set for 40 minutes. Typical analytical chromatogram representing the guaifenesin as starting compound, methocarbamol as an end product with β-isomer of methocarbamol as a potential process-related impurity was shown in Figure 3a.
Preparative HPLC conditions
An Agilent prep-HPLC system equipped with a quaternary pump (1,200 series), auto-sampler (5,000 μl loop capacity), PDA-detector, and preparative fraction collector system (1,200 series) was used. The chromatographic data were analyzed using “Chemstation” software (Agilent). A Kromasil-100 octadecyl column (250 mm length with 21.2 mm diameter and 10-μm particle size) was used for loading the sample. An analytical method was established in the linear gradient mode of separation to resolve the impurity and the parent compound. The same chromatographic conditions were scaling up for prep-HPLC to isolate and purify the required impurity fractions. The eluent (mobile phase) was composed of 0.1% v/v aqueous formic acid as solvent-A and ACN as solvent-B in the ratio of 95:5 (v/v) respectively was used in the preparation chromatographic method. The flow rate: 3.2 ml/min, injection volume: 1,000 μl was set. The eluents were monitored at 274 nm during the chromatographic run time of 40 minutes. Approximately, 10 mg/ml of sample was prepared using 5 ml methanol as a diluent to load into the column. The typical preparative chromatogram was shown in Figure 3b.
 | Figure 3. (a). Typical analytical chromatogram representing the retention time (minute) of guaifenesin (23.759), methocarbamol (29.973) as process-related impurities and β-isomer of methocarbamol (26.332) as the analyte of interest (b). Typical preparative chromatogram representing the retention time (minute) of β-isomer of methocarbamol (31.995) as the analyte of interest. [Click here to view] |
RESULTS AND DISCUSSION
Detection of impurities by HPLC
The representative analytical chromatogram of methocarbamol as the parent compound and its β-isomer as a potential process-related impurity was achieved by using the HPLC method and is depicted in Figure 3a. The β-isomeric impurity of methocarbamol under present investigations was eluted at a retention time of about 26.332 minutes, while methocarbamol eluted at about 29.973 minutes and guaifenesin was eluted at 23.759 minutes.
Isolation of the impurities by prep-HPLC
A simple RP-HPLC system, detailed under the heading “Preparative HPLC conditions” was applied for isolating the process-related impurity. Under these chromatographic conditions, methocarbamol eluted at about 31.995 minutes, whereas the chromatographic run time was about 40 minutes. The impurity portion was collected between 30.0 and 33.0 minutes of eluents. The respective fractions were concentrated by evaporating the ACN portion under vacuum in normal ambience, on a rotavapor (Buchi, Model# R124). The targeted impurity present in the aqueous layer was extracted using methylene chloride. The aqueous layer fraction was concentrated by evaporating at room temperature under a high vacuum on a rotavapor. Before carrying out spectroscopic experiments, the purity of the impurity was examined in the analytical HPLC method and was found to be 97.2%, (Fig. 3b).
 | Figure 4. LC-MS/MS spectra of methocarbamol. [Click here to view] |
 | Figure 5. LC-MS/MS spectra of β-isomer of methocarbamol. [Click here to view] |
LC-MS/MS analysis
LC-MS/MS analysis of methocarbamol APIs and its β-isomer as a potential process-related impurity was performed using the chromatographic system as described under the heading “Mass spectrometry.” The retention time in nominal mass spectra obtained in full scan mode analysis showed that the β-isomeric impurity of methocarbamol exhibited comparable molecular ion at m/z 241 and fragmentation pattern to that of methocarbamol. The full scan mass spectra analysis showing m/z 240.90 [M-H] (Fig. 4), and 240.92 [M-H] (Fig. 5) for methocarbamol and its β-isomer, respectively, supports the above findings. Based on these findings, it was presumed that the impurity was identical to that of methocarbamol which could be a positional isomer.
Structural elucidation of process-related impurity I
1H NMR data of methocarbamol (CDCl3, Avance 300 MHz): 3.75 (s, 3H, -OCH3), 3.90 (m, 2H, -CH2 at C-10), 4.38 (m, 2H, -CH2 at C-8), 4.91 (s, 1H, -OH), 6.0 (s, 2H, -NH2), 6.92 (m, 4H, Ar-H).
1H NMR data of β- isomer of methocarbamol (CDCl3, Avance 300 MHz): 3.62 (m, 1H, -CH2 at C-10), 3.75 (s, 3H, -OCH3), 3.81 (m, 1H, -CH2 at C-10), 4.30 (m, 1H, -CH2 at C-8), 4.44 (m, 2H, -CH2 at C-8 and –CH at C-9), 4.81 (s, 1H, -OH), 6.0 (s, 2H, -NH2), 6.92 (m, 4H, Ar-H).
The spectral data of β-isomer and methocarbamol were compared. The mass spectral analysis showed molecular ion for impurity at m/z 240.9 [M-H]. It was presumed that the impurity was identical to that of methocarbamol which could be a positional isomer. The 1H NMR spectrum of methocarbamol and its β-isomer are similar except C-9 and C-10 positions. C-9 in methocarbamol is substituted by –OH group and C-10 by carbamate group, whereas, in β-isomer (impurity), groups are exchanged by which it has become positional isomer of methocarbamol.
This change in position of the groups clearly reflected and differentiated the NMR spectra of two compounds (Figs. 6 and 7). In both the compounds, a functional group interchange showed an influence in the chemical shift values of protons in C-9 and C-10 positions. When compared with methocarbamol in β-isomer chemical shift values of protons at C-10 were shifted to the lower regions (i.e. from 3.90 to 3.62), and this might be due to the interchange in neighboring groups. As evident from the spectral data findings, it was affirmed the molecular formula of the impurity as C11H15NO5 and the same was characterized as 1-hydroxy-3-(2-methoxyphenoxy) propan-2-yl carbamate.
 | Figure 6. 1H NMR spectra of methocarbamol. [Click here to view] |
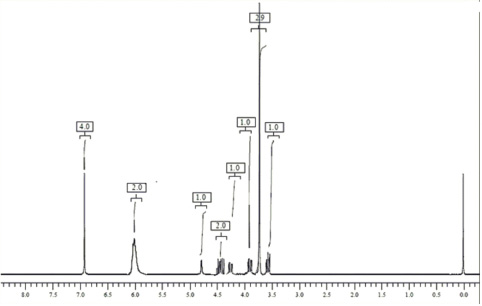 | Figure 7. 1H NMR spectra of β-isomer of methocarbamol. [Click here to view] |
CONCLUSION
The present research work describes a process-related impurity in the synthesis of methocarbamol form guaifenesin, and it was isolated using preparative LC. Based on the complete spectral analysis, the new impurity is designated as 1-hydroxy-3-(2-methoxy phenoxy) propan-2-yl carbamate and it was identified as β-isomer of methocarbamol. This research explicates the identification, isolation, and structural elucidation of a process-related impurity existing in the methocarbamol. The impurity was separated using RP-HPLC technique, further isolated this impurity by preparative LC. The isolated β-isomeric impurity of methocarbamol was identified using spectroscopic techniques. This research work demonstrated the application of NMR and MS in the efficient structural elucidation of new degradation products of bulk drugs. The formation of the impurity was also discussed in brief.
CONFLICT OF INTEREST
No conflict of interest declared.
FINANCIAL SUPPORT
None.
REFERENCES
Alessi-Severini S, Coutts RT, Jamali F, Pasutto FM. High-performance liquid chromatographic analysis of methocarbamol enantiomers in biological fluids. J Chromatogr, 1992; 582:173–79. CrossRef
Ali N, Hegazy M, Abdelkawy M, Abdelaleem E. Simultaneous determination of methocarbamol and its related substance (Guaifenesin) in two ternary mixtures with ibuprofen and diclofenac potassium by HPTLC spectrodensitometric method. J Planar Chromatogr Mod TLC, 2012a; 25:150–55. CrossRef
Ali NW, Hegazy MA, Abdelkawy M, Abdelaleem EA. Simultaneous determination of methocarbamol and its related substance (guaifenesin) in two ternary mixtures with ibuprofen and diclofenac potassium by RP-HPLC method. J Liq Chromatogr Relat Technol, 2012b; 35:2229–42. CrossRef
Desai S, Patel A, Gabhe SY. Isolation and characterization of impurities present in 8-chlorotheophylline. Indian J Pharm Sci, 2011; 73:79–84. CrossRef
Elkady EF. Simultaneous determination of diclofenac potassium and methocarbamol in ternary mixture with guaifenesin by reversed phase liquid chromatography Talanta, 2010; 82:1604–07. CrossRef
El-Yazbi FA, Amin OA, El-Kimary EI, Khamis EF, Younis SE. Simultaneous determination of methocarbamol and aspirin in presence of their pharmacopeial-related substances in combined tablets using novel HPLC-DAD method. Drug Dev Ind Pharm, 2019; 45:265–72. CrossRef
Erk N, Ozkan Y, BanoÄŸlu E, Ozkan SA, Sentürk Z. Simultaneous determination of paracetamol and methocarbamol in tablets by ratio spectra derivative spectrophotometry and LC. J Pharm Biomed Anal, 2001; 24:469–75. CrossRef
Farsam H, Souri E, Gharavi N. A new HPLC technique for the separation of methocarbamol enantiomers. J Pharm Pharmacol, 1999; 51:873–75. CrossRef
O’Neil MJ. The merck index: an encyclopedia of chemicals, drugs, and biological. Merck Sharp & Dohme Corp., Kenilworth, NJ, pp 657–60, 2006.
Raju TV, Seshadri RK, Arutla S, Mohan TSSJ, Rao IM, Nittala SR. Development and validation of a precise, single HPLC method for the determination of tolperisone impurities in API and pharmaceutical dosage forms. Scientia Pharmaceut, 2013; 81:123–38. CrossRef
Sweetman SC. Martindale: the complete drug reference. 37th edition, Pharmaceutical Press, London, UK, pp 75–6, 2011.
United States Pharmacopeial Convention. Methacarbamol, In: United States Pharmacopeia/National Formulary (USP XXI). United States Pharmacopeial Convention Inc., Rockville, MD, pp 662–3, 1984.
Walash MI, Belal F, Eid M, El-Abass SA. Spectrofluorometric determination of methocarbamol in pharmaceutical preparations and human plasma. J Fluoresc, 2011; 21:555–61. CrossRef
Walash MI, Belal F, El-Enany N, Eid M, El-Shaheny RN. Stability-indicating HPLC method with fluorescence detection for determination of methocarbamol in tablets: application to therapeutic drug monitoring. J Liq Chromatogr Relat Technol, 2012; 35:2021–41. CrossRef
Yale HL, Pribyl EJ, Braker W, Bergeim FH, Lott WA, Muscle-relaxing compounds similar to 3-(o-Toloxy)-1,2-propanediol.1 I. aromatic ethers of polyhydroxy alcohols and related compounds 2,3. J Am Chem Soc, 1950; 72:3710–16. CrossRef
Zha W, Zhu Z. Determination of methocarbamol concentration in human plasma by high performance liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 2010; 878:831–35. CrossRef