
INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disease characterized by a progressive deterioration in memory that occurs in the middle- to old-aged people (Dong et al., 2012). The condition arises from the degeneration of a significant neuronal population in the affected. The Alzheimer's Disease International—worldwide federation of Alzheimer associations, estimated that more than 50 million people were living with dementia worldwide in 2018, more than 80% of which is contributed by AD. More than 70% of this AD population was found to reside in the poor and developing countries (ADR, 2018). Despite numerous potential research studies, there is no effective treatment for the disease other than acetylcholinesterase inhibitors for the treatment of dementia symptoms. Hence, in recent years, scientists are equally focusing on the diagnostic markers for early disease detection. Diagnostic biomarkers like detecting Aβ and tau levels in cerebrospinal fluid and amyloid positron emission tomography scan helped in early detection of the disease. However, all these tests are complicated and highly expensive. Olfactory dysfunction has been identified as an early indicator of AD and is reported to occur even before the development of dementia symptoms in AD patients (Wu et al., 2013). Olfactory functions like olfactory identification and olfactory discrimination parameters are found to be affected in AD. The third function, called the olfactory threshold, is found to be less altered in the disease though reports prove a decrease in the olfactory threshold in mild cognitive impaired patients (Djordjevic et al., 2008). Many clinical data have confirmed a significant dysfunction in olfactory identification of AD patients, but reports regarding the other functions remain non-uniform to date (Devanand et al., 2010). Olfactory tests such as the University of Pennsylvania smell identification tests and Sniffin sticks aiding in olfactory diagnosis are used sometimes for AD detection in the clinical scenario. However, olfactory dysfunctions associated with other dementia types like Parkinson's disease, frontotemporal dementia pose a problem in confining these tests to only for the diagnosis of AD (Nicola, 2011). The early pathological accumulation of Aβ and tau in the olfactory bulb of affected is reported to be responsible for the olfactory dysfunction. However, the exact pathological mechanism, especially the variations in neurotransmitter systems of the olfactory system, associated with AD, is not fully understood.
Transgenic animal models are often used to study AD-related pathophysiology since they are found to mimic the disease condition in most cases naturally. Such models helped in demonstrating numerous mutations like that of the amyloid precursor protein, presenilin, and beta-secretase causing disturbances in equilibrium of Aβ production leading to the pathogenesis of AD (Klafki et al., 2006). Consequently, transgenic mouse models possessing Aβ-related mutations are used to elucidate pathological pathways for AD. Scientists used the transgenic model, AβPPswe/PS1E9 mice, for studying the olfactory dysfunction associated with AD and reported Aβ deposits and olfactory marker protein downregulation in the olfactory bulb. The animals exhibited Aβ deposition in bulb within 4 months of the study, while the deposition took almost 7 months to get detected in the hippocampal area (Wu et al., 2013). The model could successfully prove the occurrence of olfactory deficits in AD. However, this transgenic model is highly uneconomical and time-consuming. Hence, we tried to study the olfactory deficit associated with AD in a non-transgenic mice model.
The Aβ peptide, especially Aβ (1–42), is identified as one of the primary causes of AD, and the dysregulation in amyloidogenic Aβ processing has been found to develop senile plaques in the affected. Accumulation of these plaques has been found to imbalance Aβ clearance rate and turn them into dangerous toxic deposits promoting other hallmark pathologies of AD and ultimately resulting in significant neurodegeneration (Sadigh-Eteghad et al., 2015). External administration of Aβ peptide in mice brains via i.c.v injection using a stereotactic apparatus was found to induce behavioral abnormalities and also the Aβ pathophysiology associated with AD. This technique is beneficial as it lends controllability to AD investigation and makes it possible for the researchers to assume that the pathological changes arise solely from Aβ administration and not from any other factors, including individual differences that occur in non-pathogenic models of AD (Kim et al., 2016). The present study is designed to evaluate the olfactory dysfunction occurring in AD and to study the neurochemical changes responsible for it in mice brains post different intervals of i.c.v Aβ injection. Moreover, the olfactory data were compared to the memory data and correlated with neurotransmitter variations in the olfactory bulb and hippocampus tissues, providing preliminary evidence to the synaptic changes leading to these deficits.
MATERIALS AND METHODS
Materials
All the reagents used in the study were of analytical grade. Human Aβ (1–42) peptides were procured from Anaspec biotechnology, USA and processed as per the instructions. Other major chemicals like 2′,7′-dichlorofluorescein diacetate (CAS Number: 4091-99-0), Cresyl violet stain (CAS Number: 10510-54-0), GABA (CAS Number: 56-12-2), serotonin (CAS Number: 50-67-9), and L-glutamic acid standards (CAS Number: 56-86-0) for neurotransmitter estimations were procured from Sigma-Aldrich, Bengaluru, India. Ellman's reagent (CAT number: 22582) was purchased from Thermo Fisher Scientific, Bengaluru, India.
Methods
Animals
The animal experiments were conducted according to the regulation of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA). The experiment protocol was approved by the institutional animal ethical committee (IAEC No: JSSCP/199/2016), and adequate care was taken to minimize pain for the animals during the study.
2–3-months-old female C57BL/6 mice were procured from a CPCSEA approved breeder. The animals were housed at the quarantine facility of JSS College of Pharmacy, Mysuru. Animals were provided with adequate food and water facilities and kept under normal temperature conditions of 23°C ± 2°C. 12-hour light/dark cycle was also appropriately maintained. Female mice have been used for the study since women have been reported to have a higher AD incidence rate than men. Scientific evidence also suggests an increased expression of Apolipoprotein E4, the major genetic risk to AD, in females compared to males (Vina and Lloret, 2010).
Experiment protocol
Post-acclimatization period, C57BL/6 female mice were prepared for the study. The animals were maintained at constant temperature and humidity conditions (22°C–23°C and 40%) and were allowed limited access to food and water. Only the subjects that weighed within ±5% deviation from the average were included for the study. The subjects were divided into normal, sham control, and Aβ injected groups. The sham control group received phosphate-buffered saline (PBS) intracerebroventricularly. The Aβ injected group was divided into three different subgroups for studying the behavioral and neurochemical parameters at different time points (7, 17, and 28) post the i.c.v injection. The 7th-day group comprised of animals after 7 days of i.c.v injection, the 17th-day group consisted of animals after 17 days from the single i.c.v injection, and the 28th-day group accommodated the last set of animals after 28 days from the single i.c.v injection. The behavioral assessments (olfactory and memory) were recorded separately for on the 7th, 17th, and 28th day.
i.c.v administration of Aβ (1–42)
The peptide obtained commercially was treated with pre-chilled 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to dissolve any aggregates. The solution was then sonicated, incubated, and evaporated to remove any traces of HFIP. 1 μM of Aβ stock was then prepared in dimethylsulphoxide and diluted to tenfold (100 μM) in PBS to develop the Aβ monomer solution. The oligomers were prepared by incubating the monomer solution for 5 days at 37°C, and care was taken to provide a humid environment (Kim et al., 2016). The aggregated form of Aβ (1–42) was administered by i.c.v injection using a stereotaxic apparatus (Steolting, USA) consisting of a 28-gauge stainless-steel needle of 3.0 mm length (Hamilton). Briefly, mice were anesthetized with intraperitoneal (i.p.) injection of xylazine (20 mg/kg) and Ketamine (80 mg/kg) cocktail and placed in a stereotaxic frame. The needle was inserted unilaterally 1 mm to the right of the midline equidistant from each eye and 1 mm posterior to a line drawn through the anterior base of the eyes. The injection volume of 5 μl of Aβ (1–42) was delivered gradually using the following coordinates from bregma: anteroposterior = −0.1 mm, mediolateral = 1 mm, and dorsoventral = −3 mm (Souza et al., 2016). The sham-operated mice received 5 μl of PBS. Special care was taken to maintain the animal body temperature.
The Aβ dose used in the study was obtained from the literature. However, a separate set of animals were studied for the experimental concentration at 3 and 5 μl volumes for 7 days as a pilot study. The 5 μl injected group exhibited better memory dysfunction compared to the 3 μl group and hence, considered for further studies. The doses mentioned of all chemicals used in i.c.v Aβ administration was found to cause no animal mortality but still induce AD.
Behavioral parameters
Olfactory measurements
The behavioral assessment of experimental animal’s olfaction was carried out according to the Yang and Crawley buried pellet and habituation/dishabituation test protocols with slight modifications.
Buried pellet test
Buried pellet test measures the overall olfactory ability of animals, necessary for their foraging and social activities. It determines the capability of an overnight fasted animal to uncover a buried food pellet. The mice were prior familiarized to a food pellet (cheese) to confirm their palatability. The mice were then placed in clean cages with bedding of 3 cm depth and allowed to familiarize for about 5 minutes (Yang and Crawley, 2009). A food pellet was buried about 1 cm below the surface in a random corner of the cage. Time animal took to uncover the food pellet was recorded in seconds. The mice were considered to have uncovered the pellet when they started eating it.
Habituation/Dishabituation test
Habituation/dishabituation test determines the animal’s ability to distinguish between odors. The test involved the presentation of different food odors to mice in a sequence of social/familiar (banana flavor) and non-social/novel odors (chocolate/fruit flavors). The test was performed in two steps, which involved pre-test acclimatization and then the analysis for habituation and dishabituation (Yang and Crawley, 2009). In the pre-test acclimatization, the mice subjects were placed in a testing cage and were allowed to acclimatize for 30 minutes. A clean, dry applicator with familiar smells was inserted into the cage through a hole to acclimatize the animals with these smells. The presentation order for all odors was determined during each test phase. Animals were presented first with plain water solution, followed by the familiar odor and lastly, either one of the novel odors. The applicator was dipped in the odor solution for 2 seconds and then inserted into the cage. A stopwatch was used to record the time spent by the subjects sniffing the applicator tip.
Memory assessment by passive/inhibitory avoidance test
The behavioral assessment of mouse learning and memory was carried out according to the Quillfeldt’s step-down latency protocol with slight modifications. Inhibitory/passive avoidance test is usually performed using a step-down apparatus which teaches animals to avoid their innate tendencies to escape an aversive stimulus. A typical step-down apparatus consisted of an illuminated box with a frontal sliding door 0.1 cm steel grids which are almost 1 cm apart. The study was divided into two phases: a training and test phase. In the training phase, the mice were diligently taught that stepping down with the four paws on to the grid will receive prompt shock on their feet. Animals received a foot shock of 0.2–1.0 mA for 3.0 seconds. In the test phase, the animals were subjected to the same conditions as in the training phase but without the delivery of a foot shock. A ceiling of 180 seconds was maintained, and the step-down latency was measured to assess the degree of memory retention (Quillfeldt, 2006).
Animal euthanasia and preparation of samples
The 7th-day and the 17th-day group of animals were euthanized immediately after assessment of their behavior data on the respective days. The normal, sham control, and the 28th-day group of animals were sacrificed immediately after evaluation of behavioral parameters on the 28th day. The brains were excised and washed thoroughly in ice-cold PBS and normal saline. The olfactory bulb and hippocampal tissues were immediately separated and stored in −80°C from the brains other than for recording whole brain weight and histopathological analysis. The separated tissues were processed further for reactive oxygen species (ROS), acetylcholinesterase, and neurotransmitter estimations.
Brain weight determination
Whole mice brains were immediately removed from the skull and weighed carefully in a digital balance and brain weights recorded in wet weight (g).
Reactive oxygen species determination
ROS determination was carried out as per the protocol described by Tang et al. (2015) with slight modifications. DCFH-DA reagent was used to measure the generation of intracellular ROS. DCFH-DA gets converted into fluorescent DCF upon reaction with ROS. The mice brain tissues (olfactory bulb and hippocampus) were isolated and homogenized with 0.9% sodium chloride solution to produce 1:9 (g/ml) homogenates. This homogenized solution was then centrifuged at 100 g for 10 minutes at 10°C. The supernatant was collected, and 2 μl of the solution added into 198 μl of Phosphate buffer saline (PBS pH 7.5). Furthermore, 100 μl from the prepared solution was mixed with 100 μl of DCFH-DA solution in a 96-well microplate. The ROS levels were detected using a fluorescent microplate spectrophotometer (Perkin, Elmer) at an excitation wavelength of 485 nm and an emission wavelength of 525 nm.
Acetylcholinesterase estimation
Acetylcholinesterase levels were determined by Ellman’s method with slight modifications. The bulbar and hippocampal regions from each group were removed, weighed, and homogenized separately in 0.03 M sodium phosphate buffer (pH-7.4) to make 10% w/v homogenate. 0.4ml aliquots of the homogenate was added to a cuvette containing 2.6 ml phosphate buffer (0.1 M, pH 8) and 100 μl of 5,5′-Dithiobis (2-nitrobenzoic acid) (Ellman’s reagent) The contents of the cuvette were thoroughly mixed and absorbance measured at 412 nm using a UV–visible spectrophotometer (Shimadzu, Japan) was recorded as the basal reading. When absorbance reached a stable value, 20 μl of acetylthiocholine iodide was added as a substrate to initiate the reaction. Absorbance change was recorded for 10 minutes at 30 seconds intervals. One unit of acetylcholinesterase activity was defined as the number of micromoles of acetylthiocholine iodide hydrolyzed per min per mg of protein. Acetylcholinesterase activity expressed in μmol/minute/mg (Palit et al., 2015). The protein concentration of tissue homogenates was determined using the Bradford assay.
Neurotransmitter estimation
Measurement of serotonin levels
Serotonin measurement in mice brains was performed using HPLC-EC method (Maheswari et al., 2019). The brain samples were homogenized with 0.9% perchloric acid and centrifuged at 10,000 rpm for 15 minutes. The supernatant was collected and filtered through a 0.22-μm filter and used for High-Performance Liquid Chromatography (HPLC) analysis. Mobile phase composed of 82.4% water, 17.6% methanol, 20 mM of disodium hydrogen orthophosphate, 15 mM of citric acid, 9 mM DL10-camphor sulfonic acid, 1.512 Mm of triethylamine, and 0.0876 mM of ethylene diamine tetraacetic acid (EDTA) with a flow rate of 0.7 ml/minute. HPLC system with an electrochemical detector (Model 1645, Waters, USA) and C-18 column (Sphericob, RP C18, 5 μm particle size, 4.6 mm × 250 mm at 30ºC) was used for the analysis. Serotonin standard was injected separately. The neurotransmitter concentration in samples was calculated by comparing with the standard peak area and expressed in ng/mg of wet tissue.
Measurement of glutamate and gamma aminobutyric acid (GABA)
Tissues were homogenized in 500 μl of deionized water and centrifuged at 10,000 rpm for 20 minutes. Supernatants were filtered through a 0.22-μm filter. 10 μl of the filtrate was diluted in 990 μl of deionized water for HPLC analysis. The mobile phase comprised of 0.1 M monosodium phosphate and 0.5 mM EDTA dissolved in 23% methanol and diluted with distilled water. The solution pH was adjusted with 1 M phosphoric acid. The amino acids were derivatized by ortho-phthalaldehyde (OPA)/sodium sulfite solution in ethanol (22 mg of OPA dissolved in 500 μl of absolute ethanol, 500 μl of 1 M sodium sulfite, and 900 μl of 0.1 M sodium tetraborate buffer, pH adjusted to 10.4 using 5 M sodium hydroxide) (Monge and Fornaguera, 2009). HPLC system with an electrochemical detector (Model 1645, Waters, USA) and C-18 column (Maheswari et al., 2019) (Sphericob, RP C18, 5 μm particle size, 4.6 mm × 250 mm at 30ºC) was used for the analysis. GABA and L-glutamic acid standard solutions were separately injected. Sample neurotransmitter concentrations were calculated by comparing with the peak area of standard and expressed in μg/mg of wet tissue.
Histopathological analysis by cresyl violet staining
The isolated whole brain samples were immediately transferred into 10% formalin solution. The formalin-fixed samples were cut into coronal sections of 3–5 μm. Sections were then deparaffinized with two xylene washes and hydrated with descending grades of alcohol and distilled water. All the slices were stained with 0.5% Cresyl Violet solution for 3–10 minutes and rinsed immediately with distilled water (Wang et al., 2011). The prepared slides were differentiated in 95% ethyl alcohol for 2–10 minutes and dehydrated using absolute alcohol. The sections were then mounted in permanent mounting medium and observed for Nissl’s granules. The neuronal count was measured using Olympus MagVision software (calibrated with micrometer). All the brain samples were captured at 40× magnification and subjected for counting (number of neurons/100 μm).
Statistical analysis
Statistical analysis was performed using GraphPad Prism (version 6.0 software). Analysis of Variance (ANOVA) was used for comparison followed by appropriate post-hoc tests (Tukey’s/Bonferroni’s). p-value ≤ 0.05 was considered as statistically significant.
RESULTS AND DISCUSSION
Effect of i.c.v administration of Aβ (1–42) on behavioral parameters
Olfactory parameters
Early olfactory dysfunction has been reported in AD. More than 80% of AD patients are found to exhibit hyposmia/anosmia (partial/complete olfactory loss) even before memory dysfunction occurs in an AD patient (Doty, 2008). Odor identification deficits are reported to predict the clinical conversion of mild cognitive impairment to AD. Studies on transgenic AβPP/PS1 mice also correlated to the clinical findings (Wu et al., 2013). In the present study, olfactory dysfunction was measured in female C57BL/6 mice using two different olfactory assays: Buried pellet test and Habituation/Dishabituation test. Unlike humans, animals use the sense of smell for their livelihood and all social activities. Therefore, it is easier to assess olfactory parameters in rodents.
Buried pellet test measures the overall olfactory capacity of an animal. We assessed the olfactory function of the animal using its ability to unbury a piece of deeply placed food (cheese) pellet from a thick bedding layer. The results from the study were in correlation with the previous reports of olfactory dysfunction associated with AD. Animals took significantly increased time to uncover the pellet after 7, 17, and 28 days of i.c.v injection compared to the respective normal groups [115.66 ± 4.97 vs. 45.5±2.78; 118.52±7.04 vs. 50.5±5.26, and 95.67 ± 8.11 vs. 50.00 ± 4.31 (seconds)] post i.c.v injection (p < 0.001) (Fig. 1a). However, the unburying time decreased (p < 0.05) in the 28th day compared to 7th- and 17th-day groups.
Habituation to an odor is the progressive decrease in the olfactory investigation (sniffing) towards a repeated odor presentation. Dishabituation is the reinstatement of sniffing when a novel odor is presented. Therefore, the habituation/dishabituation test is primarily used to evaluate the animal’s olfactory distinguishing ability between social (familiar) and novel odors (Yang and Crawley, 2009). In the present study, we recorded the sniffing time of animal separately for familiar (banana flavor) and novel (chocolate/fruity) odors, after presenting the animals with plain water. The Aβ injected animal took significantly increased sniffing time for the novel scent but interestingly, we also found that the diseased animals took significantly increased sniffing time even for the prior habituated smell compared to the normal groups (p < 0.001) [Fig. 1b(i)]. However, the 28th-day group of animals showed a significantly decreased sniffing time (42.50 ± 4.71 seconds) for familiar smell compared to the 7th- and 17th-day group of animals [42.50 ± 4.71 vs. 58.66 ± 2.43 and 57.50 ± 4.11 (seconds), respectively] (p < 0.05). The sniffing time for novel smells significantly increased on the 7th, 17th, and 28th days compared to sham control groups [94.5 ± 4.67 vs. 19.6±2.21, 97.50 ± 8.61 vs. 27.66 ± 5.33, and 98.67 ± 5.21 vs. 27.66 ± 4.87 (seconds), respectively] (p < 0.001) [Fig. 1b(ii)]. The findings from the assay not only showed the loss of olfactory discrimination ability but also indirectly indicated the loss of the animal’s odor identification capacity since the subjects could not also identify the familiar smell.
.png) | Figure 1. Effect of i.c.v Aβ injection on olfactory and memory parameters. (a) Buried pellet test, (b) olfactory habituation/dishabituation test for i) familiar odor ii) novel odor, and (c) step down latency by passive avoidance test. All values were expressed as Mean ± SEM, n = 6. Data were analyzed by two-way ANOVA followed by post-hoc Bonferroni test. (***p < 0.001, ###p < 0.001, and ##p < 0.01 compared to the Aβ groups with normal and sham control, respectively. &p < 0.05 and $p < 0.05 compared the 7th-day group to 17th-day groups, respectively). [Click here to view] |
Memory parameter
Passive avoidance test helps in assessing the learning and memory responses in animals. Step down latency is a good indicator of retention performance/reference memory of animals (Quillfeldt, 2006). The step-down latency of the 7th-day, 17th-day, and 28th-day groups significantly decreased compared to their respective normal groups [25.66 ± 4.78 vs. 180.00 ± 10.40, 33.50 ± 6.99 vs. 178.50 ± 5.33, and 60.66 ± 4.21 vs. 180.00 ± 7.97 (seconds)] (p < 0.001) (Fig. 1c). However, the 28th-day subjects showed a significant increase in step down latency compared to the 7th-day group (p < 0.05). We propose that the cognitive deficits observed in the study are a result of the loss of olfactory functions. In normal conditions, mice extensively use their olfactory functions when subjected to cognitive tasks. The results obtained from our olfactory tests and previous studies indicated a significant olfactory loss associated to AD, which eventually exacerbates the Aβ-induced cognitive impairment subsequently leading to poor memory and learning responses and increased confusion in the affected animals. Most previous reports have studied the i.c.v effects of Aβ until the 7th day of injection (Souza et al., 2016), indicating to the acute effects of the unilaterally administered toxin on brain functions. The improved performance of 28th-day group could be due to the decreasing influence of Aβ (1–42) on hippocampal and olfactory synaptic functions as discussed in the forthcoming result sections.
Effect of i.c.v Aβ (1–42) on whole brain weight
Magnetic resonance imaging (MRI) studies of AD brains indicated global brain atrophy and reduced cerebral volumes. The neurodegeneration induced by AD could be considered responsible for the brain atrophy accompanying the disease (Scahill et al., 2003). Unaffordability of animal MRI setups led us to the determination of wet brain weight of animals. Any changes in the brain weight could indirectly indicate to neurodegeneration/atrophy. The whole-brain of animals was excised immediately and weighed to determine the wet brain weight. Correlating to the clinical reports, the Aβ-treated groups showed significantly decreased total brain weight compared to the normal group on the 7th, 17th, and 28th days [1.020 ± 0.088 vs. 0.714 ± 0.0660, 0.712 ± 0.0320, and 0.7110 ± 0.0530 (g)] (p < 0.05) post-i.c.v injection (Fig. 2).
Effect of i.c.v Aβ (1–42) on ROS levels in the mice olfactory bulb and hippocampus
ROS highly contributes to the free radical population inducing oxidative stress. Brain and its structures are comparatively more prone to oxidative stress majorly because of its higher lipid content and paucity of anti-oxidant enzyme levels compared to other organs (Morrison and Lyketsos, 2005). Aβ is reported to augment oxidative stress in AD conditions and thereby facilitate tau expression in the affected brains. Transgenic AβPP/PS1 mice were measured for mitochondrial dysfunction using ROS, membrane potential, and cytochrome c oxidase activity in the limbic and cortical regions. Hippocampus exhibited the highest ROS levels along with other parameters, and these levels were proportional to the mitochondrial Aβ levels. The degree of cognitive impairment was also found to be in correlation with the mitochondrial Aβ levels, indicating to the role of ROS in Aβ pathology of AD (Dragicevic et al., 2010) In this study, we analyzed the ROS levels in the olfactory bulb and hippocampal tissue of female black mice using DCFH-DA assay. i.c.v injection of Aβ significantly increased the ROS levels in the olfactory bulb in 7th-, 17th-, and 28th-day groups compared to the normal and sham control group (p < 0.01) as represented in Table 1. As reported in the transgenic model studies, the hippocampal tissue also expressed higher levels of ROS compared to the normal group (Table 1). The increased ROS levels in the study indicated probable mitochondrial dysfunction in the olfactory bulb and hippocampus of the i.c.v injected animals.
.png) | Figure 2. Effect of i.c.v Aβ (1–42) injection on brain weight. All values are expressed as Mean ± SEM, n = 5. Data were analyzed by one-way ANOVA, followed by post-hoc Tukey’s multiple comparison test. *p < 0.05 compared to the normal group. [Click here to view] |
Effect of i.c.v administered Aβ (1–42) on acetylcholinesterase enzyme levels of bulbar and hippocampal tissues
Acetylcholinesterase (AChE) enzyme metabolizes acetylcholine, primary neurotransmitter for learning and memory functions, into acetate and choline groups. Thereby it is an indirect indicator of acetylcholine (ACh) levels. Cholinergic deficits play an important role in the etiopathogenesis of AD, and AChE inhibitors are the only FDA approved drugs for deterring dementia symptoms of AD. The olfactory bulb possesses extensive cholinergic innervations and is essential for olfactory learning and processing. Reports have confirmed that the cholinergic system plays an important role in modulating olfactory behaviors, especially olfactory discrimination (Ogg et al., 2018). Aggregation of Aβ oligomers is reported to compromise acetylcholine neurotransmission and thereby cause neurotransmitter imbalances. Aβ has been found to complex with AChE and potentiate the neurodegenerative effects induced by the peptide (Grimaldi et al., 2016). In this study, AChE levels were estimated by Ellman’s method. The Aβ treated animals significantly increased the AChE levels in the olfactory bulb and hippocampus tissues compared to the normal and sham control groups (Table 2). The olfactory bulb AChE levels of 7th-,17th-, and 28th-day groups significantly increased compared to the normal group [p < 0.05; 17.51 ± 0.740 vs. 24.56 ± 0.96, 25.21 ± 1.34, and 23.02 ± 1.69 (mM/minute/mg)]. The AChE level in the olfactory bulb of the 28th-day group did not show any significant difference with the other groups while the levels significantly increased compared to the normal hippocampal tissue (29.43 ± 0.87 mM/minute/mg) (p < 0.05).
.png) | Table 1. Effect of i.c.v injection of Aβ (1–42) on bulbar and hippocampal ROS levels. [Click here to view] |
.png) | Table 2. Effect of i.c.v injection of Aβ (1–42) on AChE levels of olfactory bulb and hippocampus. [Click here to view] |
Effect of i.c.v Aβ (1–42) on neurotransmitter levels Olfactory bulb and Hippocampus
Neurotransmitters are primary chemical messengers necessary for proper neuronal conduction. They are essential for maintaining synaptic and cognitive functions. Neurotransmitters reportedly associated with AD are ACh, glutamate, GABA, and serotonin. As discussed in the previous section, ACh is undoubtedly the most prominently affected neurotransmitter in this disease condition. Glutamate, the primary excitatory neurotransmitter, is found to induce AD by causing excitotoxicity in neurons. Glutamate transport is found to be affected in AD, causing accumulation of glutamate at synapse leading to excitotoxicity and neuronal death. However, Aβ has been found to inhibit the N-methyl-D-Aspartate receptor functions (Revett et al., 2013). i.c.v Aβ(1-42) has been found to decrease the duration of high-frequency long-term potentiation in rat brains, the dimeric and larger forms responsible for inhibitory effects (Cullen et al., 1997) Glutamate is found to be essential for normal olfactory functioning and a reduction in glutamate levels is reported to have adverse effects on olfactory processing (Harvey and Heinbockel, 2018). GABA, the primary inhibitory neurotransmitter, is found to be essential for proper memory functions, dendritic outgrowth, and synaptogenesis in the hippocampus (Li et al., 2016). GABA is also found to be vital in modulating neural plasticity network within the olfactory system by regulating the differentiation of bulbar interneurons necessary for the consolidation of new odor memories (Akiba et al., 2009). GABA levels were found to reduce in the temporal cortex of AD patients. Transgenic AD mice models have also portrayed diminished GABAergic neuronal functions in AD (Li et al., 2016). Serotonin is yet another neurotransmitter which is found to be affected in AD condition. Serotonin plays an essential role in the brain by regulating functions necessary for social activities. Serotonin (5HT) levels are found essential for proper mood regulation and memory functions. Studies have reported beneficial effects of serotonergic activation in AD (Gaudry, 2018). Chronic administration of 5HT4 agonists and acute administration of Selective serotonin reuptake inhibitors have found to slow down amyloid pathology and decrease Aβ production, respectively (Schiavone et al., 2017). 5HT4 agonists are nootropics benefitting memory and cognitive functions and additionally are found to induce ACh release (Claeysen et al., 2015). The exact role of 5HT in olfactory processing is still not well understood. Unlike other neurotransmitters, serotonergic innervations to the olfactory bulb are supplied through centrifugal fibers from the raphe nucleus in the brain stem and not present intrinsically. Nevertheless, long-term 5HT modulation of olfactory sensory neurons by sustained raphe activation is found to increase odor discrimination function in-vivo (Gaudry, 2018).
.png) | Table 3a. Effect of i.c.v injection of Aβ (1–42) on bulbar and hippocampal levels of serotonin. [Click here to view] |
Neurotransmitter estimations were performed by HPLC-EC method with a difference in the mobile phase used. Aβ injection significantly decreased the serotonin levels compared to the normal group both in the hippocampus and olfactory bulb of the female black mice (p < 0.05, Table 3a). The Glutamate levels in the 7th and 17th day of hippocampus significantly increased compared to the normal group (p < 0.05, Table 3b) while significantly decreased in the olfactory bulb of the Aβ-treated groups. The GABA levels of olfactory bulb significantly reduced on the 7th and 17th day compared to the normal group (p < 0.05). All results were in correlation with the previous reports. As discussed earlier, cognitive functions are moderated by neurotransmitters. The increase in glutamate levels of hippocampus probably destroyed synaptic functions and affected the cognitive levels. The decreased Glutamate and GABA in olfactory bulb levels likely contributed to the olfactory discrimination loss of Aβ injected animals and overall olfactory dysfunction. The decrease in glutamate and AChE levels in 28th-day groups compared to the normal groups correlated to the olfactory and memory parameters.
Effect of i.c.v Aβ (1–42) on bulbar and hippocampal neurodegeneration by cresyl violet staining
Neurodegeneration is a significant feature of AD, and its extent was ascertained in the present study by staining Nissl granules using cresyl violet dye. Nissl granules are stacks of the rough endoplasmic reticulum (with ribosomes) and help in protein synthesis (Farlex medical dictionary). It indicates the neuronal status in a disease condition. In the present study, bulbar and hippocampal neurodegeneration was measured by Nissl’s/cresyl violet staining. The neuronal count was measured. The neuronal count of bulb and hippocampus decreased significantly on the 17th day after i.c.v injection compared to normal groups [86.33 ± 23.10 vs. 395.67 ± 30.74 (p < 0.01), 38.67 ± 4.50 vs. 67.67 ± 4.36 (p < 0.05)] (Fig. 3c). Significant neurodegeneration exhibited in both the tissues correlated with the ROS and neurotransmitter levels. However, the Aβ administered olfactory bulb tissues significantly reduced in neuronal count compared to the normal tissues. The decrease in the neuronal count after i.c.v injection in both the tissues, prominently in the olfactory bulb, could probably be responsible for the reduced mice brain weight indicated in the whole brain weight parameter of this study. The neurodegeneration, along with neurochemical parameters of olfactory bulb and hippocampus justified the olfactory dysfunction and cognitive deficits post i.c.v injection.
.png) | Table 3b. Effect of i.c.v injection of Aβ (1–42) on Glutamate and GABA levels of olfactory bulb and hippocampus. [Click here to view] |
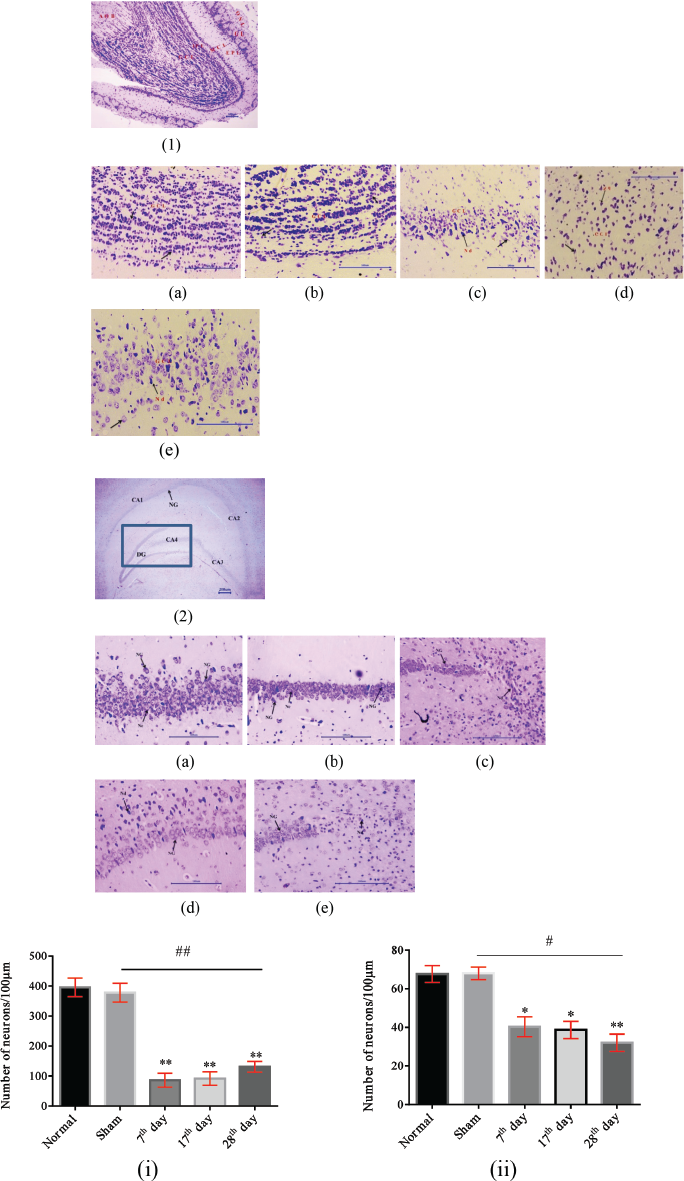 | Figure 3. Effect of i.c.v injection of Aβ (1–42) on the bulbar and hippocampal neuronal count by cresyl violet staining. a. (1) 10× image of the normal olfactory bulb (a) 40× image of the normal bulb, (b) 40× image of sham control bulb, (c) 40× image of 7th-day olfactory bulb post i.c.v injection, (d) 40× image of 17th-day olfactory bulb post i.c.v injection, and (e) 40× image of 28th-day olfactory bulb post i.c.v injection. b. (2)10× image of the normal hippocampus, the CA4 and DG sections marked. (a) 40× image of the hippocampus, (b) 40× image of sham control hippocampus, (c) 40× image of 7th-day hippocampus post-i.c.v injection, (d) 40× image of 17th-day hippocampus post-i.c.v injection, and (e) 40× image of 28th-day hippocampus post i.c.v injection. c. Neuronal count of cresyl violet stained tissues. (i) Olfactory bulb and (ii) Hippocampus. All values are expressed as Mean ± SEM, n = 3. Data were analyzed by one-way ANOVA, followed by post-hoc Tukey’s multiple comparison test. *p < 0.05, **p < 0.01 compared to normal group, #p < 0.05 and ##p < 0.01 compared to the sham control group. [Click here to view] |
CONCLUSION
Findings from this study confirm the occurrence of a prominent olfactory deficit in a non-transgenic AD mice model. The study also provided additional information on olfactory disturbances caused by Aβ peptides. Intracerebroventricular administration of Aβ (1–42) induced olfactory dysfunction from the 7th-day post i.c.v injection. However, the deficits (both olfactory and memory) started to slowly reduce in the 28th-day group, possibly indicating to a decrease in Aβ accumulation induced by the acute pathogenic model. ROS levels in the olfactory and hippocampal tissues related to the neurodegeneration occurred. The neurotransmitter disturbances associated with memory and olfaction were in correlation with the recorded behavioral changes. The study succeeded in providing preliminary evidence for the neurochemical changes in the olfactory bulb, causing olfactory dysfunction in AD. However, further studies on other parts of the olfactory system are required to establish the exact olfactory correlation in AD. Moreover, expression studies of important synaptic markers in the olfactory bulb are essential to prove the neurochemical pathophysiology of olfactory dysfunction associated with the neurodegenerative disease.
ACKNOWLEDGMENTS
Ms. Anjali Raj is thankful to the Department of science and technology for providing INSPIRE fellowship (IF160640) during the study period. The authors are also grateful to JSS Academy of Higher Education and research for providing necessary infrastructure facilities.
CONFLICT OF INTEREST
The authors declare no conflict of interest for this manuscript.
FINANCIAL SUPPORT
Nil.
REFERENCES
ADR. 2018. Available via https://www.alz.co.uk/research/world-report-2018 (Accessed 26 June 2019).
Akiba Y, Sasaki H, Huerta PT, Estevez AG, Baker H, Cave JW. Gamma-Aminobutyric acid-mediated regulation of the activity-dependent olfactory bulb dopaminergic phenotype. J Neurosci Res, 2009; 10:2211–21. CrossRef
Claeysen S, Bockaert J, Giannoni P. Serotonin: a new hope in Alzheimer’s disease? ACS Chem Neurosci, 2015; 7:940–3. CrossRef
Cullen WK, Suh YH, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by β-amyloid precursor protein fragments. Neuroreport,1997; 8:3213–7. CrossRef
Devanand DP, Tabert MH, Cuasay K, Manly J, Schupf N, Brickman AM, Andrews H, Brown TR, DeCarli C, Mayeux R. Olfactory identification deficits and MCI in a multi-ethnic elderly community sample. Neurobiol Aging, 2010; 9:1593–600. CrossRef
Djordjevic J, Jones-Gotman M, De Sousa K, Chertkow H. Olfaction in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging, 2008; 5:693–706. CrossRef
Dong S, Duan Y, Hu Y, Zhao Z. Advances in the pathogenesis of Alzheimer's disease: a re-evaluation of amyloid cascade hypothesis. Transl Neurodegener, 2012; 1:18. CrossRef
Doty RL. The olfactory vector hypothesis of neurodegenerative disease: is it viable? Ann Neurol, 2008; 63:7–15. CrossRef
Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J Alzheimers Dis, 2010; Suppl 2:S535–50. CrossRef
Gaudry Q. Serotonergic modulation of olfaction in rodents and insects. Yale J Biol Med, 2018; 1:23–32.
Grimaldi M, Marino SD, Florenzano F, Ciotta MT, Nori SL, Rodriquez M, Sorrentino G, D'Ursi AM, Scrima M. β-Amyloid-acetylcholine molecular interaction: new role of cholinergic mediators in anti-Alzheimer therapy? Future Med Chem, 2016; 11:1179–89. CrossRef
Harvey JD, Heinbockel T. Neuromodulation of synaptic transmission in the main olfactory bulb. Int J Environ Res Public Health, 2018; 15:2194. CrossRef
Iuliana N-A. Olfactory dysfunctions in Alzheimer’s disease, the clinical spectrum of Alzheimer's disease. In: De La Monte S (ed.). The charge toward comprehensive diagnostic and therapeutic strategies. IntechOpen, Europe, pp 127–44, 2011.
Kim HY, Lee DK, Chung BR., Kim HV, Kim Y. Intracerebroventricular injection of amyloid-β peptides in normal mice to acutely induces Alzheimer-like cognitive deficits. JoVE, 2016; 109:1–6. CrossRef
Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J. Therapeutic approaches to Alzheimer's disease. Brain, 2006; 11:2840–55. CrossRef
Li Y, Sun H, Chen Z, Xu H, Bu G, Zheng H. Implications of GABAergic neurotransmission in Alzheimer’s disease. Front Aging Neurosci, 2016; 8:1–12. CrossRef
Maheswari DU, Anand T, Manu TM, Khanum F, Sharma RK. Motion sickness induces physiological and neuronal alterations in a mouse model. IJPSR, 2019; 10:1650–9.
Monge-Acuña AA, Fornaguera-Trías J. A high-performance liquid chromatography method with electrochemical detection of gamma-aminobutyric acid, glutamate and glutamine in rat brain homogenates. J Neurosci Methods, 2009; 2:176–81. CrossRef
Morrison AMS, Lyketsos CG. The pathophysiology of Alzheimer’s disease and directions in treatment. ANS, 2005; 8:256–70.
Ogg MC, Ross JM, Bendahmane M, Fletcher ML. Olfactory bulb acetylcholine release dishabituates odour responses and reinstates odour investigation. Nat. Commun, 2018; 9:1868. CrossRef
Palit P, Mukherjee D, Mandal SC. Reconstituted mother tinctures of Gelsemium sempervirens L. improve memory and cognitive impairment in mice scopolamine-induced dementia model. J Ethnopharmacol, 2015; 159:274–84. CrossRef
Quillfeldt JA. Behavioral methods to study learning and memory in rats. In: Andersen ML, Tufik S (ed.). Rodent model as tools in ethical biomedical research. Springer International Publishing, Cham, Switzerland, pp 271–311, 2006. CrossRef
Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid ß peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci, 2013; 1:6–23. CrossRef
Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: a crucial factor in Alzheimer's disease. Med Princ Pract, 2015; 1:1–10. CrossRef
Scahill RI, Frost C, Jenkins R, Whitwell JL, Rossor MN, Fox NC. A longitudinal study of brain volume changes in normal aging using serial registered magnetic resonance imaging. Arch Neurol, 2003; 7:989–94. CrossRef
Schiavone S, Tucci P, Mhillaj E, Bove M, Trabace L, Morgese MG. Antidepressant drugs for beta amyloid-induced depression: a new standpoint? Prog Neuropsychopharmacol Biol Psychiatry, 2017; 78:114–22. CrossRef
Souza LC, Jesse CR, Antunes MS, Ruff JR, de Oliveira Espinosa D, Gomes NS, Donato F, Giacomeli R, Boeira SP. Indoleamine-2,3-dioxygenase mediates neurobehavioral alterations induced by an intracerebroventricular injection of amyloid-β1-42 peptide in mice. Brain Behav Immun, 2016; 56:363–77. CrossRef
Tang J, Yuan Y, Wei C, Liao X, Yuan J, Nanberg E, Zhang Y, Bornehag, CG, Yang X. Neurobehavioral changes induced by di (2-ethylhexyl) phthalate and the protective effects of vitamin E in Kunming mice. Toxicology Res, 2015; 4:1006–15. CrossRef
Vina J, Lloret A. Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-β peptide. JAD, 2010; 20:S527–33. CrossRef
Wang Y, Wang B, Zhu M-T, Li M, Wang HJ, Wang M, Ouyang H, Chai ZF, Feng WY, Zhao YL. Microglial activation, recruitment and phagocytosis as linked phenomena in ferric oxide nanoparticle exposure. Toxicol Lett, 2011; 205:26–37. CrossRef
Wu N, Rao X, Gao Y, Wang J, Xu F. Amyloid-β deposition and olfactory dysfunction in an Alzheimer’s disease model. J Alzheimers Dis, 2013; 37:699–712.
Yang M, Crawley JN. Simple behavioural assessment of mouse olfaction. Curr Protoc Neurosci, 2009; 48:8–24. CrossRef