
INTRODUCTION
G-protein-coupled receptors (GPCRs) are the largest group of membrane receptors and commonly found in many organisms, such as human. GPCRs regulate many signaling systems and physiological processes in human body. Therefore, GPCRs comprise the largest family of individual drug targets, accounting for approximately 19% of the established drug-targeted portions of the genome (Rask-Andersen et al., 2014). According to the global market share of therapeutic drugs, GPCRs-targeted drugs are reported to have approximately 27% market share of the total products (Hauser et al., 2018).
Dopamine receptor is one of GPCRs which has been targeted for drug development. There are many pharmacological disorders and conditions which are related to dopamine receptor, such as attention-deficit/hyperactivity disorder (ADHD), schizophrenia, Parkinson’s disease, bipolar disorder, depression, restless leg syndrome, hyperprolactinemia, pituitary tumors, hypertension, gastroparesis, nausea, and erectile dysfunction (Beaulieu and Gainetdinov, 2011; Iversen and Iversen, 2007). After the discovery of dopamine as a putative independent neurotransmitter in the nervous system more than 50 years ago, research related to dopamine indexed by Pubmed was found to reach more than 100,000 published papers (Björklund and Dunnet, 2007). Also, the dopaminergic research has become one of the main focuses in modern biological psychiatry (Iversen and Iversen, 2007). For example, the relationship between dopamine and schizophrenia has been explained comprehensively by Howes and Nour (2016) and their finding was further improved recently (Nour et al., 2018). Another example, the role of dopamine in addiction was also recently discussed (Caprioli et al., 2014; Nutt et al., 2015; Solinas et al., 2018).
The dopamine receptor-related-structure-based research had to deal with the lack of structural information about the protein structures and their ligand complexes. Therefore, previous research was mainly focused on the development of homology models of dopamine receptors protein for structure-based drug design purpose (Platania et al., 2012). However, recent research data were able to reveal the crystal structures of D2-like receptors with their antagonist bound to the active sites. Chien et al., (2010), Wang et al., (2017), and Wang et al., (2018) were able to crystallize dopamine D3 (PDB code: 3PBL), dopamine D4 (PDB code: 5WIU), and dopamine D2 (PDB code: 6CM4) receptors, respectively.
Previous finding of dopamine D2 receptor enabled researchers to perform virtual screening, while the recent protein crystal structure becomes a guide for the homology modeling of dopamine D2 receptor. In this paper, we compare the protein of dopamine D2 receptor homology model from GPCRdb with the recent crystal structure of the receptor. Also, we present the virtual screening protocol validation for the newly crystallized dopamine D2 receptor.
MATERIALS AND METHODS
Comparing the full structure of proteins
Homology model of inactive state dopamine D2 receptor was retrieved from GPCRdb at www.gpcrdb.org (Pándy-Szekeres et al., 2017). X-ray crystal structure of dopamine D2 receptor [PDB code: 6CM4 (human dopamine D2 receptor in complex with risperidone)] was obtained from Protein Data Bank at www.rcsb.org (Berman et al., 2000). Homology model of dopamine D2 receptor was converted into FASTA format by using Advanced Protein Sequence Converter (APSC). The FASTA format was submitted to the Basic Local Alignment Search Tool (BLAST) protein (Altschul, 1990; Gish and States, 1993) to perform sequence similarity with 6CM4.
To obtain the deviation of atom distance between the homology model and the X-ray structure, alignment was performed by using command line align in Pymol v2.10 (Schrodinger, 2018) without further refinement. The alignment of CA atoms and backbone atoms were also performed to obtain their root mean square deviation (RMSD) for all regions. RMSD of full atoms, CA atoms, and backbone atoms were also performed for aligned regions only which were obtained from TM-align (Zhang and Skolnick, 2005).
Comparing the binding site regions of proteins
X-ray crystal structure of dopamine D2 was separated from their bound ligand by using SPORES v1.3—mode splitpdb (Brink and Exner, 2009) and mol2 structures of 6CM4 and risperidone were obtained. The binding site coordinates and the gridbox sizes were calculated based on binding site coordinates of risperidone in 6CM4 by using PLANTS v1.2—mode bind (Korb et al., 2006). The method also produced active site regions and active site amino acid residues as PLANTSactiveSite.mol2 and PLANTSactiveSiteResidues.mol2, respectively. The same procedures were applied to the aligned homology model of dopamine D2 receptor.
PLANTSactiveSiteResidues.mol2 from homology model and X-ray model were superimposed in Pymol v2.10. RMSD of each residue was calculated using command rms_cur with the assumption that atoms are stored in identical order. The visualization and RMSD calculation were performed in Pymol v2.10.
Comparing the binding pose of risperidone in both models
Risperidone was redocked into dopamine D2 receptor homology model and X-ray model. Molecular docking was performed by using PLANTS v1.2 —mode screen with 50 replications for each model. The binding site coordinates and grid box sizes were obtained from prior step. The PLANTS v.1.2 results were then further analyzed with Pyplif v0.1.1 (Radifar et al., 2013) to see the interaction fingerprinting between risperidone and amino acid residues after being redocked.
Molecular docking with PLANTS v1.2 resulted in 50 binding poses for each replication. The binding pose which gave the best ChemPLP score was extracted and compared to the actual pose from 6CM4. The actual pose from 6CM4 was also compared to the binding pose which exhibited the best TcPlif score according to Pyplif. Visualization and RMSD calculation was performed in Pymol v2.10.
Virtual screening validation of 6CM4
Virtual screening validation was performed by PLANTS v1.2. GPCR Decoy Database (GDD) and GPCR ligand database for dopamine D2 receptor antagonist were obtained from Cavasotto Lab (Gatica and Cavasotto, 2012). Preparation of ligand set (529 compounds) and decoy set (20631 compounds) were performed by Open-Babel v2.31 (O’Boyle et al., 2011) and SPORES v1.3—mode reprot. For each compound, 50 binding poses were generated with five replications. Further filtering system was performed with Pyplif v0.1.1., which produce the Tanimoto Coefficient (TcPlif) score. PLANTSactiveSiteResidues.mol2 was used as a substitution for the whole protein in Pyplif calculation. Following analysis using Pyplif, binding pose of each compound was sorted according to TcPlif score. Binding pose with the best TcPlif score was extracted and EF1% was calculated based on ChemPLP score.
RESULTS AND DISCUSSION
According to Wang et al., (2018), the newly crystallized dopamine D2 receptor (PDB code: 6CM4) is a chimeric type receptor with three thermo stabilizing mutations (I1223.40A, L3756.37A, and L3796.41A). The receptor was found to be bound to risperidone, an antagonist of D2 receptor, so that the protein is in inactive state conformation.
The sequence (Fig. 1) similarity between 6CM4 and the homology protein was 99% (with 98% coverage) and this result was an acceptable index to comply with 30% identity-rule-of-thumb (Peterson et al., 2009). However, similarity percentage between two proteins was not conclusive to show whether they are homologues. Instead, E-value and bit scores are more sensitive and reliable than percent identity for interfering homology (Pearson, 2013). The lower the E-value, the better the hit significance (E-value for same proteins is 0.0). On the other hand, higher bit score (>50) represents better alignment (Madden, 2013). NCBI BLASTP result of 6CM4 and the homology protein was 1e-128 and 548 for E-value and bit score, respectively. Therefore, it can be concluded that both proteins are homologue.
Alignment of 6CM4 and homologous protein was performed with Pymol (Fig. 2). RMSD value for all atoms, C-alpha atoms and backbone atoms in the protein structure were found to be less than 3Å after being superimposed (Table 1). Even after recalculation using the aligned region only (Xu and Zhang, 2010; Zhang and Skolnick, 2004), the RMSD were still >2 Å. However, an RMSD value of less than 3Å for homology model is still considered to be high quality (Rayan, 2009; Reva, 1998; Xie et al., 2017).
 | Figure 1. The sequence of dopamine D2 receptor homology model and X-ray crystallography model. [Click here to view] |
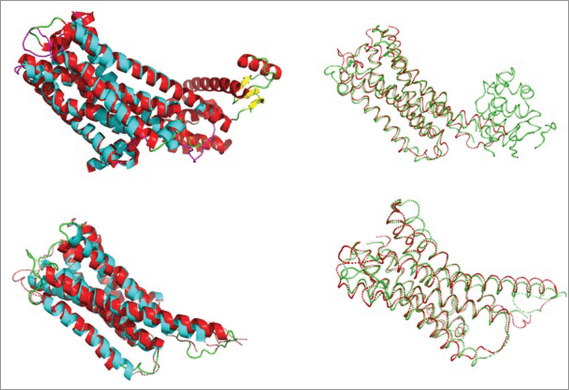 | Figure 2. Superimposed structure of all regions and aligned regions which involves full atoms and CA atoms only. [Click here to view] |
The main problem with RMSD value is that the size of the protein has become a dependent variable for RMSD distribution (Kufareva and Abagyan, 2012). Therefore, it affects the similarity between proteins (Pascual-Garcia et al., 2010). To overcome the drawback of RMSD dependency towards the protein size, TM-score was developed (Zhang and Skolnick, 2004). In this experiment, the TM-score of 6CM4 and the homology structure was found to be 0.60846 so that both of them were found in about the same fold (Zhang and Skolnick, 2005).
As the binding site is an important part in molecular docking, comparison of binding site residues were also performed. The binding site residue was generated by PLANTS (Korb et al., 2006) from the 6CM4 and the coordinates were used as a binding site. According to Wang et al., (2018), there are eight residues (Asp-114, Thr119, Phe-198, Phe-382, Trp-386, Phe-389, Thr-412, and Tyr-416) which affect the affinity of risperidone when they were mutated (Fig. 3). Homology model of dopamine D2 receptor is able to present those essential residues with RMSD < 2 Å.
The most essential residue for antagonist binding was Asp-114 which forms a hydrogen bonding with amino group in ligand (Ekhteiari Salmas et al., 2017; Kalani et al., 2004). The distance difference between ASP-114 in 6CM4 and the homology protein was 0.720Å. In addition, according to Kalani et al., (2004), risperidone antagonist will bind to the other essential residue which is Ser-197 and both proteins were able to conserve the residue within the binding site (0.864 Å apart).
There were two residues in homology protein which fall into >2 Å difference with the X-ray structure, which were Trp-100 and Tyr-408 with 6.296 and 2.837 Å distance, respectively. Tyr-408 was facing deeper into the binding site on the homology structure, while Trp-100 was facing more to the outside of the binding site.
Redocking of risperidone on 6CM4 and homology protein was performed by using the binding site coordinates from 6CM4 crystal structure. The re-docking system in both proteins failed to obtain RMSD < 2 Å when ChemPLP score was designated as a filtering system (Fig. 4). There was only 28% of the binding pose from 6CM4 which gave RMSD <2 Å and 100% of the binding pose from homology model fell with RMSD > 2 Å. Therefore, the binding pose was re-picked according to the TcPlif score which was obtained from Pyplif v.1.1. Pyplif is a python-based open source to analyze the interaction fingerprinting (IFP) between ligand and amino acid residues. This program generates IFP as a bit string value and the similarity of the binding pose is compared to the reference as a Tanimoto Coefficient (TcPlif) score.
 | Table 1. RMSD value of dopamine D2 receptor when superimposed to the X-ray crystallography model. [Click here to view] |
The use of TcPlif as a filtering system for redocking step resulted in different RMSD for 6CM4. It can be seen that binding poses selected according to TcPlif score were able to give RMSD <2 Å for all binding poses (100%). However, TcPlif score was not able to generate better poses for homology model, as 100% of RMSD is still more than 2 Å. Tyr-408, which faces deeper into the binding site, plays an important role against the difference pose between the actual pose of risperidone and redocking pose in homology protein.
Because homology model of dopamine D2 receptor failed to give the correct binding pose, virtual screening protocol validation was performed for 6CM4. The retrospective validation was performed to 529 ligands and 20,631 decoys. The parameter of retrospective validation is EF1% value which represents the early enrichment of the protocols (Jain and Nicholls, 2008). The better the EF1% value, the better the protocol predictivity for ligand identification. The protocol was developed according to the redocking step and EF1% value was calculated based on TcPlif-ChemPLP score. The protocol gave EF1% value of 6.238 with ChemPLP cutoff of −118.0. The EF1% value was slightly better when it was compared to the EF1% value of protein dopamine D3 receptor (4.4) which shared more than 80% similarity to the dopamine D2 receptor.
 | Figure 3. Amino acid residues which forms binding site region. Red color represents the homology model and green color represents 6CM4. [Click here to view] |
 | Figure 4. RMSD value of risperidone after redocking in (a) homology protein and (b) 6CM4. [Click here to view] |
CONCLUSION
The homology model of dopamine D2 receptor was able to share similar sequence and folding to the recent crystallized structure. However, it fails to give the correct binding pose of co-crystal ligand. Since the similarity between the docking pose and the actual pose is considered as an important parameter to obtain better predictivity, the incapability of the homology model to predict the correct binding pose will produce a bias result when it is used for the development of bioactive agents. Therefore, the use of recent crystallized dopamine D2 receptor (PDB code: 6CM4) is recommended for virtual screening. Also, the EF1% value of recent crystallized dopamine D2 receptor, which is better than dopamine D3 receptor, can be a reasonable reason for choosing 6CM4 as a protein model in developing bioactive agents for dopamine D2 receptor antagonists.
ACKNOWLEDGMENTS
This research was supported by Dexa Laboratories of Biomolecular Sciences (DLBS)-Dexa Medica, Indonesia.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol, 1990; 215(3):403–10.
Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev, 2011; 63(1):182–217.
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res, 2000; 28(1):235–242. Available via www.rcsb.org (Accessed 14 March 2018).
Björklund A, Dunnett SB. Fifty years of dopamine research. Trends Neurosci, 2007; 30(5):185–7.
Brink TT, Exner TE. Influence of protonation, tautomeric, and stereoisomeric states on protein-ligand docking results. J Chem Inf Model, 2009; 49(6):1535–46.
Caprioli D, Calu D, Shaham Y. Loss of phasic dopamine: a new addiction marker? Nature Neuroscience, 2014; 17(5):644–6; doi:10.1038/nn.3699
Chien EYT, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science, 2010; 330(6007):1091–5.
Ekhteiari Salmas R, Serhat Is Y, Durdagi S, Stein M, Yurtsever M. A QM protein–ligand investigation of antipsychotic drugs with the dopamine D2 Receptor (D2R). J Biomol Struct Dyn, 2017; 1102(August):1–10.
Gatica EA, Cavasotto CN. Ligand and decoy sets for docking to G protein-coupled receptors. J Chem Inf Model, 2012; 52(1):1–6.
Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat Genet, 1993; 3(3):266–72.
Hauser AS, Chavali S, Masuho I, Jahn LJ, Martemyanov KA, Gloriam DE, Babu MM. Pharmacogenomics of GPCR drug targets. Cell, 2018; 172(1–2):41–54.e19.
Howes OD, Nour MM. Dopamine and the aberrant salience hypothesis of schizophrenia. World Psychiatry, 2016; 15(1):3–4; doi:10.1002/wps.20276
Iversen SD, Iversen LL. Dopamine: 50 years in perspective. Trends Neurosci, 2007; 30(5):188–93.
Jain AN, Nicholls A. Recommendations for evaluation of computational methods. J Comput Aided Mol Des, 2008; 22(3–4):133–9.
Kalani MYS, Vaidehi N, Hall SE, Trabanino RJ, Freddolino PL, Kalani MA, Floriano WB, Kam VW, Goddard WA 3rd. The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc Natl Acad Sci, 2004; 101(11):3815–20.
Korb O, Stutzle T, Exner TE. PLANTS: Application of ant colony optimization to structure-based drug design. Ant Colony Optim Swarm Intell Proc, 2006; 4150:247–58.
Kufareva I, Abagyan R. Methods of protein structure comparison. Methods Mol Biol, 2012; 857:231–57.
Madden, T. 2013. The BLAST sequence analysis tool. In: The NCBI handbook [Internet]. 2nd edition, National Center for Biotechnology Information, Bethesda, MD, pp 427–37, 2013.
Nour M, Dahoun T, Schwartenbeck P, Adams R, Gerald TF, Coello C, Wall M, Dolan R, Howes O. The role of dopamine in processing the meaningful information of observations, and implications for the aberrant salience hypothesis of schizophrenia. Schizophr Bull, 2018; 44(Suppl 1):S385; doi:10.1093/schbul/sby018.941
Nutt D J, Lingford-Hughes A, Erritzoe D, Stokes PRA. The dopamine theory of addiction: 40 years of highs and lows. Nat Rev Neurosci, 2015; 16(5):305–12; doi:10.1038/nrn3939
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform, 2011; 3(10):33.
Pándy-Szekeres G, Munk C, Tsonkov TM, Mordalski S, Harpsøe K, Hauser AS, Bojarski AJ, Gloriam DE. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res, 2018; 46(D1):D440–6.
Pascual-Garcia A, Abia D, Mendez R, Nido GS, Bastolla U. Quantifying the evolutionary divergence of protein structures: the role of function change and function conservation. Proteins Struct Funct Bioinforma, 2010; 78(1):181–96.
Pearson WR. An introduction to sequence similarity (“homology”) searching. Curr Protoc Bioinforma, 2013; (Suppl 42); doi:10.1002/0471250953.bi0301s42
Peterson ME, Chen F, Saven JG, Roos DS, Babbitt PC, Sali A. Evolutionary constraints on structural similarity in orthologs and paralogs. Protein Sci, 2009; 18(6):1306–15.
Platania CBM, Salomone S, Leggio GM, Drago F, Bucolo C. Homology modeling of dopamine D2 and D3 receptors: molecular dynamics refinement and docking evaluation. PLoS One, 2012; 7(9):e44316.
Radifar M, Yuniarti N, Istyastono EP. PyPLIF: python-based protein-ligand interaction fingerprinting. Bioinformation, 2013; 9(6):325–8.
Rask-Andersen M, Masuram S, Schiöth HB. The druggable genome: evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu Rev Pharmacol Toxicol, 2014; 54(1):9–26.
Rayan A. New tips for structure prediction by comparative modeling. Bioinformation, 2009; 3(6):263–7.
Reva BA, Finkelstein AV, Skolnick J. What is the probability of a chance prediction of a protein structure with an rmsd of 6 Å? Fold Des, 1998; 3(2):141–7.
Solinas M, Belujon P, Fernagut PO, Jaber M, Thiriet N. Dopamine and addiction: what have we learned from 40 years of research. J Neural Transm (Vienna), 2019; 126(4):481–516; doi:10.1007/s00702-018-1957-2
The PyMOL molecular graphics system, Version 2.1.0. Schrödinger, LLC, New York, NY, 2018.
Wang S, Che T, Levit A, Shoichet BK, Wacker D, Roth BL. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature, 2018; 555(7695):269–73.
Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, Venkatakrishnan AJ, Huang XP, Dror RO, Shoichet BK, Roth BL. D4dopamine receptor high-resolution structures enable the discovery of selective agonists. Science, 2017; 358(6361):381–6.
Xie B, Sood A, Woods RJ, Sharp JS. Quantitative protein topography measurements by high resolution hydroxyl radical protein footprinting enable accurate molecular model selection. Sci Rep, 2017; 7(1).
Xu J, Zhang Y. How significant is a protein structure similarity with TM-score=0.5? Bioinformatics, 2010; 26(7):889–95.
Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins Struct Funct Genet, 2004; 57(4):702–10.
Zhang Y, Skolnick J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res, 2005; 33(7):2302–9.