
INTRODUCTION
Glecaprevir (GLE), (1R,14E,18R,22R,26S,29S)-26-tert-butyl-N-[(1R,2R)-2-(difluoromethyl)-1-{[(1-methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-13,13-difluoro-24,27-dioxo-2,17,23-trioxa-4,11,25,28-tetraazapentacyclo[26.2.1.0^{3,12}.0^{5,10}.0^{18,22}]hentriaconta-3,5(10),6,8,11,14-hexaene-29-carboxamide (Fig. 1a) is an hepatitis C virus (HCV) non-structural (NS) protein 3/4A protease inhibitor having potent pangenotypic antiviral activity (Fred et al., 2017). Unlike other antivirals, it does not inhibit human proteases, exhibits in vitro Half maximal effective concentration (EC50) values <5 nM across all major HCV Genotypes (GTs), and demonstrates <5-fold loss of activity against common GT1 variants at key resistance-associated positions of R155 and D168 to currently available NS3/4A PIs (Ahmed and Felmlee, 2015; Ng et al., 2014).
Pibrentasvir (PIB), (2S,3R)-1-[(2S)-2-{5-[(2R,5R)-1-{3,5-difluoro-4-[4-(4-fluorophenyl)piperidin-1-yl]phenyl}-5-{6-fluoro-2-[(2S)-1-[(2S,3R)-2-{[hydroxy(methoxy)methylidene]amino}-3-methoxybutanoyl]pyrrolidin-2-yl]-1H-1,3-benzodiazol-5-yl}pyrrolidin-2-yl]-6-fluoro-1H-1,3-benzodiazol-2-yl}pyrrolidin-1-yl]-2-{[hydroxy(methoxy)methylidene]amino}-3-methoxybutan-1-one (Fig. 1b) is a novel and pan-genotypic HCV NS5A inhibitor with EC50 values ranging from 1.4 to 5 pM against HCV replicons containing NS5A from genotypes 1–6 (Ng et al., 2017).
 | Figure 1. Structure of (a) GLE and (b) PIB. [Click here to view] |
The treatment with the United States Food and Drug Administration (USFDA) approved the combination of GLE and PIB, the directly acting antivirals for 12 weeks, resulted in a high rate of sustained virologic response in patients with stage 4 or 5 chronic kidney disease and HCV infection (Gane et al., 2017). Furthermore, this drug regimen had a favorable safety profile in previously treated or untreated patients with chronic HCV genotypes 1, 2, 4, 5, or 6 infections and compensated cirrhosis (Forns et al., 2017).
Literature survey through standard databases showed only a handful reported analytical method available for the simultaneous estimation of the drug combination with stability indicating Reverse Phase-High Performance Liquid Chromatography (RP-HPLC) method. Understanding the fact that for the routine analysis of the quality attributes of the combination in pure and pharmaceutical dosage form, an easy, reproducible, precise, accurate, and economic method must be developed to overcome the present challenge, this study is conducted. The aim of the present work is to develop validated stability indicating RP-HPLC method for simultaneous estimation of GLE and PIB in bulk and tablet formulation.
MATERIALS AND METHODS
Materials
GLE and PIB were obtained as a gift sample from SL Drugs and Pharmaceuticals Ltd., Hyderabad. The marketed formulation MEVYRET® containing 100 mg of GLE and 40 mg of PIB was purchased from GNH India Suppliers. All the chemicals and solvents used in this experimental study were of HPLC grade. The HPLC study was performed using the Waters® 2695 system with PDA detector 2996 on a reverse phase Denali C18 column of 150 mm × 4.6 mm dimension, 5 μm particle size. The system was equipped with EMPOWERS v.2 software with a manual rheodyne injector (20-μl loop).
Chromatographic conditions
The mobile phase potassium dihydrogen orthophosphate buffer (pH 4.8): acetonitrile in the ratio of 60:40 v/v was eluted at a flow rate of 1 ml/minute with an ambient column temperature 30°C. The eluate was monitored at 260 nm.
Preparation of analytical solutions
Preparation of 0.01-N potassium dihydrogen orthophosphate buffer
An accurately weighed 1.36 g of potassium dihydrogen orthophosphate was added to a 1,000 ml of volumetric flask containing 900 ml of milli-Q water. The content was further degassed by sonicating and finally, the volume was made up with distilled water. Nearly, 1 ml of triethylamine was added to maintain the pH to 4.8 with the dilute orthophosphoric acid solution.
Preparation of mobile phase
A mixture of above-prepared buffer was mixed with acetonitrile in the ratio of 60:40 v/v. The solution was further degassed by sonicator for 5 minutes and filtered through 0.45 μm under vacuum filtration.
Diluent preparation
Water and acetonitrile were mixed in the ratio of 50:50 v/v and used as the diluent for the standard and sample preparation.
Standard preparation
Accurately weighed quantities of GLE (25 mg) and PIB (10 mg) were added to a 25-ml clean dry volumetric flask and filled ¾th volume with the diluent. The contents were further sonicated for 5 minutes and the final volume was made up with the diluents to produce 1,000 ppm of GLE and 400 ppm of PIB. Afterward, 1 ml from the resultant two stock solutions was pipetted out into a 10-ml volumetric flask and the volume was made up to 10 ml with the diluents to produce 100 ppm of GLE and 40 ppm of PIB.
Sample preparation
Five tablets were accurately weighed and the average weight was computed. The weight equivalent to a tablet was transferred into a 100-ml volumetric flask and half filled with the diluent. The content was further sonicated for the duration of 25 minutes and suitably filtered to produce 1,000 ppm of GLE and 400 ppm of PIB. Afterward, 1 ml of the filtered solution was pipetted out into a 10 ml of volumetric flask and the volume was made up to 10 ml with the diluents to produce 100 ppm of GLE and 40 ppm of PIB.
Method validation
The method was validated as per the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines Q2A and Q2B, in compliance with the FDA guidance and by United States Pharmacopoeia (USP).
Linearity and range
For the estimation of linearity, six concentrations of the target analytical concentration of the formulation were taken: 25–150 μg/ml for GLE and 10–60 μg/ml for PIB. The solutions were prepared in the diluent and equivalent volumes of the solution were injected under the specific condition of chromatography. The linearity graph was plotted between the obtained average areas versus the concentration volume. The linearity of the study was expressed according to the regression coefficient value (r2) (Sawale et al., 2017).
Accuracy
The recovery (accuracy) was worked out by continuously spiking the standard drug concentrations at 50%, 100%, and 150% of the target concentration. The experiment was carried out in a triplicate manner and the data were expressed as % recovery ± confidence interval with calculated % relative error on the basis of definite and anticipated concentrations (Deodhe et al., 2017a).
Precision
The precision of the analytical method was determined through the obtained relative standard deviation (RSD) values. The inter-day variability was estimated by injecting the standard solution at three concentrations of 50%, 75%, and 150% six times in a single day, while the intra-day variability was conducted similarly on three different days (Deodhe et al., 2017b).
Robustness
The consequences of intentional alteration in the systems suitability parameters such as mobile phase composition by ±5% v/v (i.e., 65:35 % v/v and 55.45 % v/v), flow rate by ±0.1 ml/minute (i.e., 0.9 and 1.1 ml/minute), and column temperature by ±5°C (i.e., 25°C and 35°C) were thoroughly studied while maintaining the additional chromatographic conditions constant (Jha et al., 2017).
Systems suitability parameters
The study involved the estimation of reproducibility attributes of the system by determining the imperative parameters such as TPs, retention time, peak area, and tailing factor (TF) by injecting five times the standard solution (Prakash et al., 2018).
Limit of detection and quantification
Limit of detection (LOD) may be defined as the lowest concentration that can be detected by the method but not always necessary to quantify in exact value (Perumal et al., 2014).
The LOD was determined by the following formula:
LOD = 3.3 (σ/S)
where σ = standard deviation of response and S = slope of the calibration curve. The slope S may be estimated from the calibration curve of the analyte.
Limit of quantification (LOQ) may be defined as the lowest concentration that can possibly be quantified dependably with a particular level of accuracy and precision (Bauer et al., 2014).
The LOQ is determined by the following formula:
LOQ = 10 (σ/S)
where σ = standard deviation of response and S = slope of the calibration curve. The slope S may be estimated from the calibration curve of the analyte.
Degradation studies
Acid degradation studies
The 0.5 mg of the combination was taken in a 100-ml volumetric flask and diluent was added to half the volume (50 ml), sonicated for 15 minutes, and the volume was made up to 100 ml. By employing the magnetic stirrer, the content was stirred for the period of 30 minutes and centrifuged suitably at 3,000 rpm for 5 minutes. Nearly, 5 ml of the produced solution was taken, equivolume solution of 2-N HCl was added, and the content was boiled on a water bath for 1 hour. The boiled content was cooled, neutralized with the same quantity of 2-N NaOH, and the volume was made up to mobile phase to produce 100 ml. Filtration was performed through the 0.45 μm pore size nylon membrane and 20 μl volume of sample was injected into the HPLC system to record the chromatogram.
Alkali degradation studies
The 0.5 mg of the combination was taken in a 100-ml volumetric flask and diluent was added to half the volume (50 ml), sonicated for 15 minutes, and the volume was made up to 100 ml. By employing the magnetic stirrer, the content was stirred for the period of 30 minutes and centrifuged suitably at 3,000 rpm for 5 minutes. Nearly, 5 ml of the produced solution was taken, equivolume solution of 2-N NaOH was added and the content was boiled on a water bath for 1 hour. The boiled content was cooled, neutralized with the same quantity of 2-N HCl, and the volume was made up to mobile phase to produce 100 ml. Filtration was performed through the 0.45 μm pore size nylon membrane and 20 μl volume of sample was injected into the HPLC system to record the chromatogram.
Neutral degradation studies
The stress under the neutral conditions was studied by taking the combination equivalent to 0.5-mg weight and refluxing them under distilled water for 6 hours. Nearly, 5 ml of the content was taken and diluted with mobile phase to produce the resultant solution 100 ml. The content was filtered through 0.45 μm pore size nylon membrane and 20 μl volume of sample was taken for investigation.
Oxidation degradation studies
The oxidative stress offered to the drug combination was estimated by taking the 0.5-mg equivalent weight in a volumetric flask and dissolving in H2O2 (5 ml). The content was further boiled for the duration of 1 hour and kept at the room temperature to initiate the degradation. The content was then diluted, sonicated, and the volume was made up to 100 ml. The supernatant was centrifuged, filtered, and the sample of 20 μl volume was injected into the HPLC system to obtain the chromatogram.
Dry heat degradation studies
For the estimation of the thermolytic effect on the drug formulation (combination) under moderately high temperature, 0.5 mg equivalent of the combination was taken and thoroughly exposed to heat 70°C ± 1°C for 1 hour. The content was diluted with acetonitrile, stirred for half an hour, sonicated for 15 minutes, and the volume was made up to 100 ml. The above content was centrifuged at 3,000 rpm for the 5-minute duration, filtered, and 20-μl volume sample was injected in the chromatographic system.
Photo stability studies
For ultraviolet-induced degradation, 0.5 mg equivalent of the combination was taken in a petri dish and forced degraded for 3 days continuously under ultraviolet radiation of 254-nm wavelength. The radiation exposed powdered material was relocated to a volumetric flask containing the mobile phase. The above content was further sonicated for the period of 15 minutes and the volume was made up to 100 ml. The prepared solution was stirred for 30 minutes and filtered through a 0.45-μm membrane filter. Nearly, 20 μl volume of drug solution was injected into the HPLC system.
RESULTS AND DISCUSSION
Method development and optimization of chromatographic conditions
This new method was developed based on the previously reported RP-HPLC simultaneous estimation methods. From the literature, it was suggested to employ reverse phase C18 stationary phase having attributes of 250 × 4.6 mm i.d., particle size 5 μm. Therefore, the Denali C18 column was utilized for the development of chromatographic separation technique. To design and develop a method that can achieve a practical scale of multiple drug separation, the composition of the mobile phase was thoroughly learned. The selection of the mobile phase was made on the basis of the number of TPs, peak symmetry, and peak purity index, after a number of trials using binary eluant systems like methanol, phosphate buffer, acetonitrile, water, etc. pH was suitably altered in the range of 3.0–6.0 to ensure and gain desirable separation. In addition, a low pH creates an environment that prevents peak tailing and improves the ruggedness of the method. The pH was skillfully altered within 2 units of the pKa so as to assure unionization of the analyte. A pH higher than 2 units higher on the pKa often results in ionization of the analyte. Sometimes, the selection of a basic pH results in complete dissolution of the silica from the reverse phase columns. Furthermore, there is a change that both the drugs get degrade in high basic surroundings.
The elution with methanol: buffer KH2PO4 (50:50) presented a low intensity and high tailing peak. Similarly, the concentration ratio of 10:90 v/v resulted in fronting issues. When no satisfactory results were obtained, the buffer was replaced by water. However, when used with water at a ratio of 20:80 v/v, no sharp peak was detected and was found misfit for elution purpose. When the amount of water was reduced, a little betterment was observed but the peaks were not acceptable. Logically, restoring the KH2PO4 buffer (pH 4.8), when employed with acetonitrile in the ratio of 80:20 v/v, a marked improvement in the peak shape and tailing were observed. On further optimization of the ratio to 60:40 v/v, sharp peak with least tailing and high TPs were detected. The elution was performed using Denali C18 column maintained at ambient temperature with mobile phase KH2PO4 buffer (pH 4.8): acetonitrile (60:40 v/v) optimized in isocratic mode at a flow rate of 1 ml/minute in a 10-minute run-time, keeping detector at 260 nm. The retention time of GLE and PIB was found to be 2.13 and 3.46 minutes, respectively (Fig. 2a). Ideally, elution of the solutes in the time range of 3–4 minutes is considered optimum within a 10-minute run of the chromatogram. The developed method presented a similar appearance of peaks in the chromatogram in the ideal range with better resolution.
Method validation
Linearity and range
Excellent linearity was observed over the range 25–150 μg/ml for GLE and 10–60 μg/ml in the case of PIB (Table 1). The linear regression equation and regression coefficient value of GLE was found to be y = 755x + 15,780 (r2 = 0.999) and 6,900x + 2,150 (r2 = 0.999) for PIB, respectively, which correspond to highly acceptable degree of linearity (Fig. 3).
Accuracy
The recovery data for accuracy studies were calculated using the calibration curve in which the slope and Y-intercept of the graph play an imperative role in the estimation of % recovery. The developed method holds % RSD values within the acceptance limit of ±2%, i.e., 1.2%, 0.63%, and 0.29% at three different concentrations for GLE. For PIB, % RSD values of the proposed method lie below the pharmacopeia limits: 0.43%, 0.57%, and 0.75%, which indicates good accuracy of the method. The recovery data are described in Table 2.
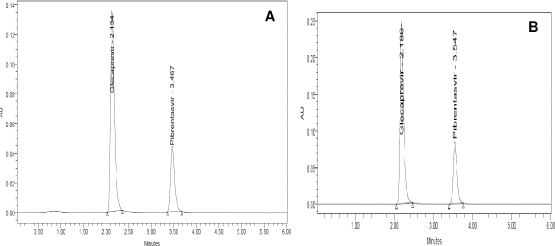 | Figure 2. Chromatogram for GLE and PIB (a) after method optimization and (b) tablet sample solution. [Click here to view] |
Precision
The method was found to be highly precise over the range of 50%–150% of the standard drug. The % RSD values were found to be less than 2% in both cases. In intra-day variability, the % RSD was found to be in the range 0.22%–0.57% (Table 3) and in inter-day variability, the % RSD range was 0.32%–0.94% (Table 4). This concluded that the variability was found to be minimal and the method is precise enough to determine the drug. The intra-day and inter-day variability or precision data are given in Tables 3 and 4.
Robustness
As the chromatographic conditions were altered intentionally, negligible changes were monitored. When the flow rate was changed from 1 to 0.9 ml/minute, the observed retention peak at 2.13 minutes for GLE and 3.46 minutes for PIB get increased to 2.38 and 3.87 minutes, respectively. When the flow rate was increased to 1.1 ml/minute, the retention time was reduced and noticed at 2.14 and 3.48 minutes for GLE and PIB, respectively. The variation of column temperature from 30°C to 35°C resulted in a shift of retention time to 2.16 minutes for GLE and 3.47 minutes for PIB (Table 5). Similarly, the swing of column temperature from 30°C to 25°C causes an increase of retention time to 2.18 minutes for GLE and 3.63 minutes for PIB. In the purposeful modification of mobile phase ratio of 65:35 v/v, the retention time was transformed to 2.21 minutes for GLE and 3.69 minutes for PIB. Likewise, the retention time was adapted to 2.19 minutes for GLE and 3.47 minutes for PIB, when mobile phase composition was changed to 55:45 v/v.
System suitability parameters
The system suitability parameters highlighted that the proposed method has enough competence and capability to be employed for regular analysis. As per the minimum requirements of monographs of USP, the developed method has the ability to give reproducible results. For GLE, the system exhibited a mean of 2,554 TPs, which is more than the minimum pharmacopeia limit of 2,000, which represents better separation, resolution, and high column efficacy. The average retention time (Rt) of GLE was recognized at 2.13 minutes. The TF of 1.43 signified good peak symmetry.
Quite similarly, the PIB system displayed a mean TP of 8,027, a value greater than the pharmacopeia limit, thereby representing enhanced column efficacy and excellent separation. The retention time (Rt) of PIB on average was perceived at 3.46 minutes. The TF of 1.27 signified good peak symmetry. The TF value of 1.27 implying no tailing and is investigative of the fact that the asymmetric factor is also equal to 1, reflecting the shape of an ideal Gaussian peak where both the factors are equal in magnitude. On the whole, it may be concluded that the developed method has the attributes of accuracy, precision, robustness, reproducibility, and can be applied for routine analysis. The system suitability parameters are depicted in Table 6.
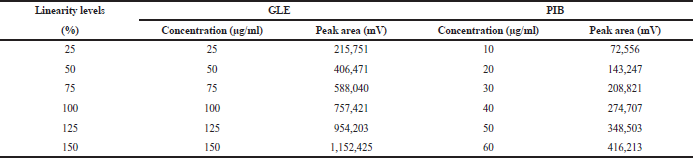 | Table 1. Linearity study of GLE and PIB. [Click here to view] |
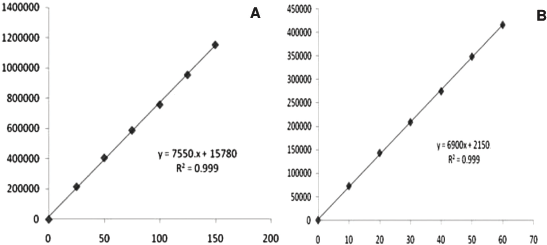 | Figure 3. Linearity plot of (a) GLE and (b) PIB. [Click here to view] |
Limit of detection and quantification
The LOD for GLE and PIB was found to be 0.21 and 0.29 μg/ml, respectively. The LOQ or GLE and PIB were found to be 0.61 and 0.89 μg/ml, respectively. These results indicate that the proposed method has a very good tendency to detect the lowest concentration of both the drug simultaneously from the formulation.
Forced degradation studies
The forced degradation study highlighted that both the drugs (GLE and PIB) were quite resistant to degradation under acidic, basic, neutral, oxidative, UV, and thermal conditions. Under all the forced conditions, no main degraded product(s) were observed in the spectra. Only under the acidic, alkaline, and oxidative stress conditions, a single degradant peak was seen predominantly in the retention time 1.38 minutes with less than 5% of the total chromatogram area (Fig. 4). The prominent appearance of the peak was observed under the oxidative stress, followed by alkaline and acid conditions with no abrupt change in the drug peaks under all the conditions. However, the stability of the drugs is profoundly dependent on pH, buffer concentration, and environmental impact. The peak of 1.38 minutes was not perceived under the neutral degradation, UV exposure, and thermal stress conditions. The plausible reason for the alkaline degradation may be the abstraction of the proton in a mechanistic way by the OH− radical. The bonding between the carbon atom and the weakly acidic alpha proton was cleaved by the strong base.
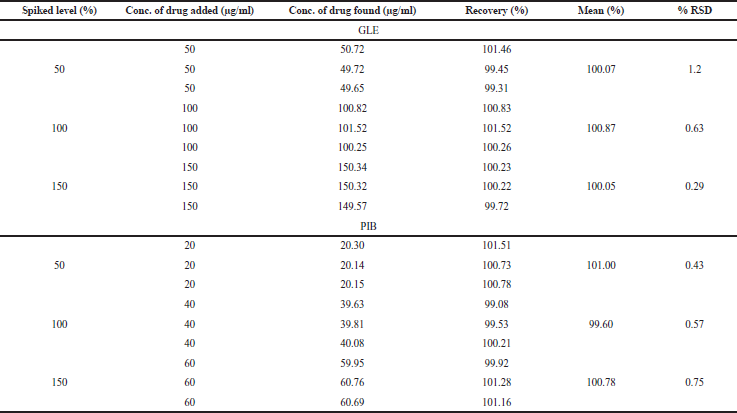 | Table 2. Recovery for accuracy studies for the combination. [Click here to view] |
.png) | Table 3. Precision data of intra-day variability. [Click here to view] |
.png) | Table 4. Precision data of inter-day variability. [Click here to view] |
.png) | Table 5. Robustness study of GLE and PIB. [Click here to view] |
.png) | Table 6. Systems suitability parameters. [Click here to view] |
Comparison with already reported methods
On comparing the analytical methods previously developed by several research groups across the globe, it was observed that the current simultaneous method possess much better attribute which will be of practical application interest. The method developed by Sreeram and Venkateswarlu (2018), where C8 column was made into the application, presented a narrow elution of solutes at 2.107 minutes (PIB) and 2.341 minutes (GLE), respectively, which was overcome by this present method quite successfully (difference of 1.5 minutes) by utilizing the C18 column. In a similar way, the produced elution technique for both the drugs with multiple components: methanol, triethanolamine, and acetonitrile (55:25:20 v/v) by Saradhi et al. (2018) was also effectively minimized by employing more greener chemicals such as buffer (pH 4.8) and acetonitrile in a low ratio of 60:40 v/v. Analogously, the reported stability data by Kumar and Raja (2018) showed a single degraded component at 2.67 minutes under all the treated conditions of acid, base, heat, oxidant, neutral, and sunlight. The re-done study truly corrected the observations by certainly distinguishing the degraded materials (at 1.38 minutes) under only three conditions (acidic, alkaline, and oxidative) from the obtained chromatogram. Finally, with comparison of the chromatographic work (0.5-mM orthophosphoric acid buffer: acetonitrile in the ratio of 75:25 v/v, pH 4.3) on the similar subject by Babu et al. (2018), a fast elution of the solutes has been found with 60:40 v/v ratio in our developed method (2.134 minutes for GLE and 3.467 minutes for PIB) along with exhibition of some contrasting forced degradation results under various conditions of heat, light, and nature.
.png) | Figure 4. Force degradation studies of GLE and PIB under (a) acid condition, (b) alkaline condition, (c) neutral condition, (d) oxidative stress, (e) UV-radiation, and (f) thermal condition. [Click here to view] |
CONCLUSION
The USFDA approved GLE and PIB combination can now be simultaneously estimated in both bulk and pharmaceutical dosage form from the developed RP-HPLC analytical method using the mobile phase system KH2PO4 buffer (pH 4.8): acetonitrile in the ratio of 60:40 v/v. The validated stress degradation studies highlighted the possible degraded components after subjecting to various stressed conditions, which will be of great concern for the chemists in a daily analysis. The results and attributes of the validated method were found to be within the prescribed limits given by ICH, which ensured adequate reproducible characteristics. This simple, accurate, precise, and economical method can be applied in the routine simultaneous analysis of bulk product and tablets.
CONFLICT OF INTEREST
The authors have declared that they have no conflict of interest.
FINANCIAL SUPPORT
None.
REFERENCES
Ahmed A, Felmlee DJ. Mechanisms of hepatitis C viral resistance to direct acting antiviral. Viruses, 2015; 7:6716–29. CrossRef
Babu D, Chetty CM, Mastanamma SK. A new force indicating RP-HPLC method development and validation for the simultaneous estimation of Pibrentasvir and Glecaprevir in bulk and its tablet dosage form. Pharm Methods, 2018; 9(2):79–86. CrossRef
Bauer LC, Santana DD, Macedo MD, Torres AG, Souza NE, Simionato JI. Method validation for simultaneous determination of cholesterol and cholesterol oxides in milk by RP-HPLC-DAD. J Braz Chem Soc, 2014; 25(1):161–8. CrossRef
Deodhe ST, Dhabarde DM, Kamble MA, Mahapatra DK. Novel stability indicating RP-HPLC method for the estimation of Pinaverium bromide in tablet formulation: assay development and validation. Eur J Anal Chem, 2017a; 12(2):3–16. CrossRef
Deodhe ST, Dhabarde DM, Kamble MA, Mahapatra DK. Development and validation of a novel stability indicating RP-HPLC method for the estimation of Entecavir in tablet formulation. Eur J Anal Chem, 2017b; 12(3):223–35. CrossRef
Gane E, Lawitz E, Pugatch D, Papatheodoridis G, Bräu N, Brown A, Pol S, Leroy V, Persico M, Moreno C, Colombo M. Glecaprevir and Pibrentasvir in patient with HCV and severe renal impairment. New Engl J Med, 2017; 377:1448–55. CrossRef
Fred P, Franco F, Armen A, Mark SS, Robert WR, Charles SL. Glecaprevir and Pibrentasvir for 12 weeks for hepatitis C virus genotype 1 infection and prior direct-acting antiviral treatment. Hepatology, 2017; 66(2):389–97. CrossRef
Jha SK, Bhaskaran S, Kamble MA, Mahapatra DK. A novel RP-HPLC based assay for the estimation of Tramadol HCl content in tablets: development and validation. Invent Impact Pharm Anal Qual Assur, 2017; 2017(4):142–6.
Kumar KR, Raja S. Simultaneous assay of two antiviral agents, Pibrentasvir and Glecaprevir, using stability indicating RP-HPLC method in bulk and tablets. Der Pharmacia Lett, 2018; 10(8):33–47.
Ng T, Reisch T, Middleton T, Mc Daniel K, Kempf D, Lu L, Wang G, Jiang L, Or YS, Pilot-Matias T. ABT-493, a potent HCV NS3/4A protease inhibitor with broad genotype coverage. Poster #636 at: 21st Annual Conference on Retroviruses and Opportunistic Infections (CROI), 3–6 March 2014, Boston, MA.
Ng TI, Krishnan P, Pilot-Matias T, Kati W, Schnell G, Beyer J. In vitro antiviral activity and resistance profile of the next generation hepatitis C virus NS5A inhibitor pibrentasvir. Antimicrob Agents Chemother, 2017; 61(5):e02558–16. CrossRef
Perumal SS, Ekambaram SP, Raja S. Analytical method development and validation of simultaneous estimation of rabeprazole, pantoprazole, and itopride by reverse-phase high-performance liquid chromatography. J Food Drug Anal, 2014; 22(4):520–6. CrossRef
Prakash O, Mahapatra DK, Singh R, Singh N, Verma N, Ved A. Development of a new isolation technique and validated RP-HPLC method for Quercetin and Kaempferol from Azadirachta indica leaves. Asian J Pharm Anal, 2018; 8(3):164–8. CrossRef
Saradhi NDVR, Jat RK, Reddy MV. A new technical method for Glecaprevir and Pibrentasvir in combined dosage forms using non polar HPLC. World J Pharm Med Res, 2018; 4(12):173–8.
Sawale V, Dhabarde DM, Mahapatra DK. Development and validation of UV spectrophotometric method for simultaneous estimation of Olmesartan Medoxomil and Chlorthalidone in bulk and tablet. Eur J Anal Chem, 2017; 12(1):55–66. CrossRef
Sreeram V, Venkateswarlu C. Stability indicating RP-HPLC method for the simultaneous estimation of glecaprevir and pibrentasvir in drug product. J Pharm Sci Res, 2018; 10(11):2757–61.
Forns X, Lee SS, Valdes J, Lens S, Ghalib R, Aguilar H, Felizarta F, Hassanein T, Hinrichsen H, Rincon D, Morillas R. Glecaprevir plus Pibrentasvir for chronic hepatitis C virus genotype 1, 2, 4, 5, or 6 infection in adults with compensated cirrhosis (EXPEDITION-1): a single-arm, open-label, multicentre phase 3 trial. Lancet Infectious Disease, 2017; 17:1062–8. CrossRef