
INTRODUCTION
The p90 ribosomal S6 kinases (RSK) is a serine/threonine kinase consisting of four closely related isoforms, i.e., RSK1, RSK2, RSK3, and RSK4; all of which have N-terminal kinase domain (NTKD), a conserved regulatory linker domain, and C-terminal kinase domain (CTKD) (Anjum and Blenis, 2008; Carriere et al., 2008; Casalvieri et al., 2017; Romeo et al., 2012; Sulzmaier et al., 2016). The RSKs are involved in activating a range of substrates responsible for cell cycle control, motility, and cell survival and are a part of the Ras/Raf/MEK/ERK pathway, which is widely overexpressed in many types of cancer (Casalvieri et al., 2017). The RSKs are activated by sequential phosphorylation at CTKD, linker domain, and NTKD (Casalvieri et al., 2017). The deregulation of RSK signaling was indicated in pre-neoplastic progression to neoplastic disease (Davies and Dunn, 2011; Reipas et al., 2013). In spite of high sequence homology (73%–80% amino acid identity), each RSK isoform can promote specific biological function. For example, RSK1 is primarily known for its involvement in cancer cell invasion and metastasis, while RSK2 was indicated as a lead molecular target for triple-negative breast cancer (Brough et al., 2011; Larrea et al., 2009; Smolen et al., 2010; Stratford et al., 2012; Reipas et al., 2013). Thus, RSK1 and RSK2 deserve important anticancer target. Reports have recorded the developed inhibitors working on ATP-binding site of the NTKD, such as SL0101, BI-D1870, and BIX 02565, while the other irreversibly binds to ATP-binding site of CTKD, including fluoremethylketone (Casalvieri et al., 2017; Houles and Roux, 2018). Although several RSK inhibitors have been identified, several issues remain, such as high clearance, short plasma half-life, and moderate tissue distribution (Houles and Roux, 2018; Jain et al., 2015).
Jain et al. (2015) described optimization of a series of difluorophenol pyridine derivatives as RSK inhibitor by employing structure-based drug design, including crystallography, conformational analysis, and scaffold morphing. In the present study, new difluorophenol pyridine derivative was proposed using three-dimensional quantitative structure–activity relationship (3D-QSAR) model of comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA). In addition, molecular docking and 50-ns molecular dynamics (MD) simulations were conducted to study the interaction mechanism and conformational changes during the interaction of difluorophenol pyridine derivative and RSK2. The free energy calculation was also performed to confirm the reliability of CoMFA and CoMSIA model and to reveal the individual energy contribution in each complex.
MATERIALS AND METHODS
Data sets
All 21 difluorophenol pyridine series were obtained from Jain et al. (2015), in which 16 of them were grouped as training set to build models of the CoMFA and CoMSIA (Table 1). The remaining compounds were assigned as test set (superscript * in Table 1) to evaluate the built CoMFA and CoMSIA models. The inhibitory activity data which were reported in the half maximal inhibitory concentration (IC50) were transformed to the logarithmic scale pIC50 (−logIC50) to enhance the linearity of QSAR models, in which pIC50 values ranged from 5.3188 to 8.3979.
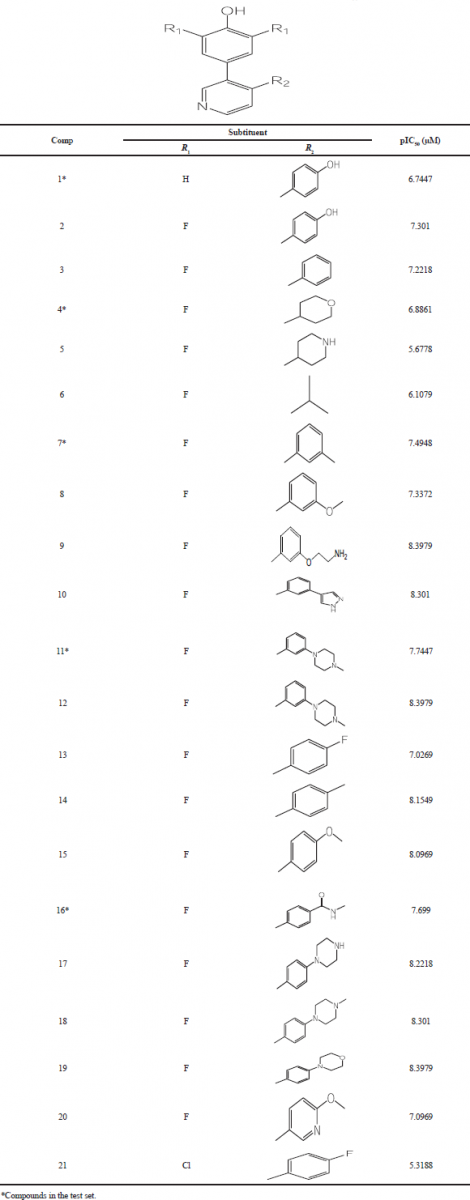 | Table 1. The structures of difluorophenol pyridine series and their pIC50 values. [Click here to view] |
SYBYL-X 2.1 (Tripos Software, Saint Louis, MO) was used to build all 3D structures of ligands. Energy minimization was performed using Tripos force field, Powell method, and Gasteiger–Huckel charges (Awasthi et al., 2018; Clark et al., 1989). The minimization was achieved when energy convergence met 0.005 kcal/mol with 1,000 cycles maximum iterations. The other topomer CoMFA parameters were left at default values except for those specifically described. As the quality of 3D-QSAR CoMFA and CoMSIA models depends primarily on molecular alignment, alignment of molecules was performed on dataset molecules based on the best active compound as the template molecule (Compound 18).
CoMFA and CoMSIA
The models of CoMFA and CoMSIA were built using SYBYL-X 2.1 (Tripos Software, Saint Louis, MO). CoMFA steric field was developed based on van der Waals interaction using Lennard-Jones potential, while CoMFA electrostatic field was built using Coulombic potential. Both CoMFA fields were generated at each lattice point in a 3D grid box of 2.0 Å in all the three directions. The energy cut-off for both steric and electrostatic fields was set to 30 kcal/mol (Wang et al., 2019; Stahle and Wold, 1988).
In the meantime, the CoMSIA steric (S), electrostatic (E), hydrophobic effects (H), and hydrogen bond donor (D) and acceptor (A) interaction fields were derived using a distance-dependence Gaussian function (Klebe et al., 1994). The same training and test set were employed to derive CoMFA and CoMSIA models. The partial least squares (PLS) algorithm with leave-one-out cross validation was employed to determine the optimal number of principal component (ONC), which was then used to correlate 3D structure of the compounds as independent variables and biological inhibitory activities (pIC50) as dependent variable (Bush and Nachbar, 1993). The cross-validation correlation coefficient of q2 value was defined as:
where, y and Å· were observed and predicted biological activities of compound i, respectively, and ȳ is the mean observed activities of the training set compounds. The value of column filtering (σ) was set to lower than 2.0 kcal/mol to reduce noise and improve efficiency. The best COMFA and CoMSIA models were judged based on high q2 and correlation coefficient (R2) values (q2 > 0.50 and R2 > 0.60), low standard error of estimate (SEE), and optimal number of component values (Golbraikh and Tropsha, 2002).
Molecular docking
The Surflex-Dock module in SYBYL-X 2.1 was employed to conduct molecular docking to investigate the binding interaction of each compound. The protein crystal structure of RSK2 complexed with LJH685 (PDB ID: 4NUS, resolution: 2.39 Å) (Aronchik et al., 2014) was imported from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (www.rcsb.org). All the water molecules and native ligand (LJH685) were removed, while polar hydrogen atoms were added to the protein. Molecular docking was performed using a protomol-based method, which was generated based on a cognate ligand and active site residues of RSK2. The compound was geometrically optimized using the Tripos force field and the Powell algorithm with a convergence criterion of 0.05 kcal/molAËš and Gasteiger–Huckel charges assigned. The compounds were automatically docked into the binding site of RSK2 with other parameters left at default values. The Surflex-Dock module generated the Total-Score, which is the representation of binding force expressed in negative logarithm of the dissociation constant (–logKd, Kd is the dissociation constant). The higher Total-Score implies stronger binding affinity, which was then selected as the docking result.
Molecular dynamics simulation and MM-PBSA calculation
The investigation of the binding interaction of ligand and protein target by molecular docking was not sufficient as docking method did not take into account receptor flexibility and water environment which are fundamental on living system. Therefore, MD simulation method was applied to resolve those issues. In the present study, MD simulation was conducted for 50 ns for Compound 18 and HA25, each complexed with RSK2. The system preparation, minimization, heating, and equilibration steps were applied as in our previous protocol (Arba et al., 2018a; 2018b). In addition, using MD trajectory, binding free energy was calculated by applying Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) approach (Kollman et al., 2000). The MM-PBSA approach was applied to 200 snapshots extracted from 30 to 50 ns. The MM-PBSA approach was described elsewhere (Arba et al., 2016; Kollman et al., 2000).
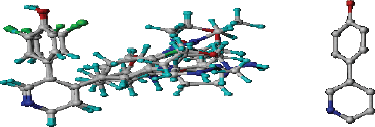 | Figure 1. 3D-QSAR structure superposition and alignment of training set (left) and common substructure used for alignment (right) of difluorophenol pyridine derivatives. [Click here to view] |
RESULTS AND DISCUSSION
The 3D-QSAR CoMFA and CoMSIA analyses were performed on 21 difluorophenol pyridine derivatives which were reported to inhibit P90 Ribosomal S6 Kinase. As the predictive ability of the QSAR model depends heavily on molecular alignment, alignment of the dataset was performed using Compound 18 as a template. The superimposition of all the compounds was done using the common skeleton as displayed on Figure 1. Furthermore, the derivation of COMFA and CoMSIA model was carried out on 16 training set compounds using PLS analysis with the result detailed in Table 2. It is showed that q2 for CoMFA and CoMSIA were 0.597 and 0.563, while R2 for CoMFA and CoMSIA were 0.993 and 0.990, respectively, which was indicative for good internal QSAR model predictability. In addition, the external predictive ability was assured using six test set compounds, which were selected based on structural diversity and biological activity. It resulted in r2pred value for CoMFA and CoMSIA was 0.996 each. In CoMFA model, steric and electrostatic accounted for 51.3% and 48.7%, respectively, while in CoMSIA model, steric, electrostatic, hydrophobic, H-bond donor and H-bond acceptor accounted for 16.2%, 34.3%, 17.0%, 22.8%, and 9.6%, respectively. The F values for CoMFA and CoMSIA were 221.414 and 153.383, respectively, while SEE values for COMFA and CoMSIA were 0.108 and 0.129, respectively. Table 3 shows the experimental and calculated pIC50 values of all the molecules, while Figure 2 displays predicted pIC50 versus observed pIC50 for training and test set compounds for both CoMFA and CoMSIA, in which almost all the points are located on the straight line.
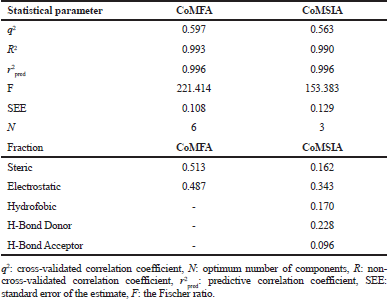 | Table 2. PLS statistic of CoMFA and CoMSIA models. [Click here to view] |
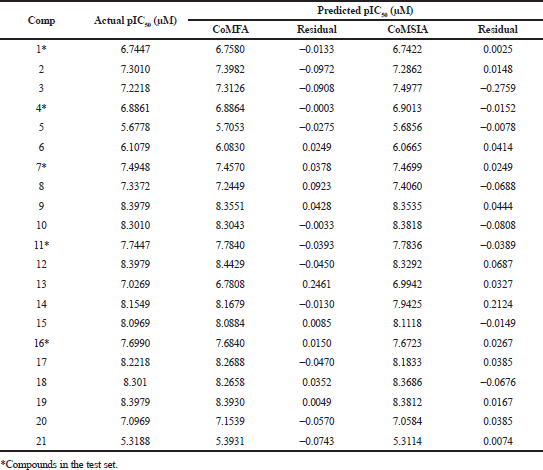 | Table 3. The experimental pIC50s and calculated pIC50s of the training and test set molecules. [Click here to view] |
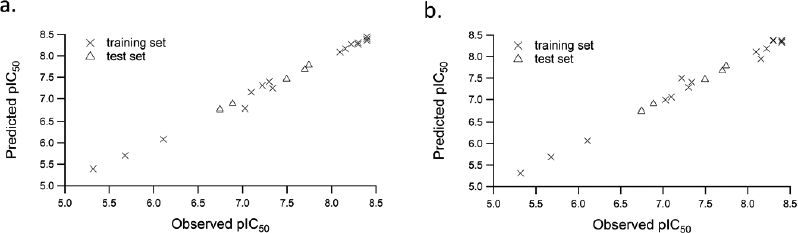 | Figure 2. Correlation of actual pIC50 versus predicted pIC50 for (a) CoMFA and (b) CoMSIA. [Click here to view] |
CoMFA and CoMSIA contour maps
The CoMFA and CoMSIA models were implemented through contour maps, which were constructed based on Compound 18 (the most active compound). The contour maps were used as guidance in designing new candidate compound as the modification in the certain area will correlate with biological activity change. The steric fields were represented by green and yellow contours, which favored and disfavored bulky groups, respectively, for enhancing inhibitory activity. As shown in Figure 3a, the presence of the green contour around piperazine ring suggested that a bulky group at this region would be favorable. The electrostatic field contour maps of CoMFA were indicated by blue and red contours, respectively (Fig. 3b). Blue contour indicates that electropositive charge will increase the activity, while red contours indicate that electronegative charge will increase the activity. The blue contours around R2 position indicate that electropositive charge would be favorable.
In the CoMSIA steric field (Fig. 4a), the green area indicates that increasing the bulky group was favorable for increasing activity, while yellow contours disfavored bulky group. The large yellow contours around R2 position indicate the unfavorable bulky group for increasing the activity. The electrostatic field of CoMSIA (Fig. 4b) is shown in blue and red contours, in which blue and red contours favored electropositive and electronegative charges, respectively, for increasing inhibitory activity. The blue contour in the piperazine ring indicates that electropositive charge substituent will increase the activity, while the red contour on the benzene ring indicates that electronegative charge substituent will increase the activity.
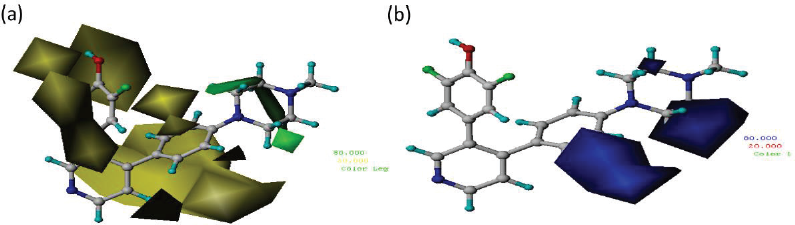 | Figure 3. CoMFA (a) steric fields: the contribution of green and yellow contours is 80% and 20%, respectively. (b) Electrostatic fields: blue and red contours are 80% and 20%, respectively. [Click here to view] |
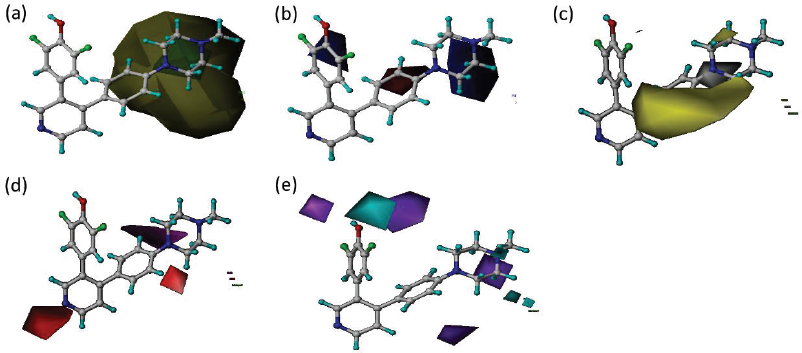 | Figure 4. CoMSIA (a) steric fields: contribution of green and yellow contours is 80% and 20%, respectively (b) electrostatic fields: blue and red contours are 80% and 20% contributions, respectively (c) hydrophobic fields: yellow and white contours are 80% and 20% contributions, respectively. (d) H-bond acceptor fields: magenta and red contours are 80% and 20% contributions, respectively (e) H-bond donor fields: cyan and purple contours are 80% and 20% contributions, respectively. [Click here to view] |
The hydrophobic field of CoMSIA model (Fig. 4c) was displayed in yellow and white contours, in which hydrophobic group was favored in yellow contours to increase the activity. The large yellow contour in the benzene ring suggests that adding hydrophobic substituents in the region may help increase the inhibitory activity.
The hydrogen bond acceptor field of CoMSIA model (Fig. 4d) is shown in magenta and red contours, in which hydrogen bond acceptor group was favorable in magenta contours for increasing the activity. The hydrogen bond donor field of CoMSIA model (Fig. 4e) is displayed in cyan and purple contours, in which hydrogen bond donor group was favored in cyan contours for increasing the inhibitory activity. The contour map of the CoMFA and CoMSIA models is useful to design new difluoro phenol pyridine derivative.
Design for new molecule
The design of new compound aims to obtain compound with increased affinity, which was performed based on Compounds 18. From the results of the study, there is a novel compound that has a higher predictive value of both predicted pIC50 CoMFA and CoMSIA than Compound 18, i.e. HA25.
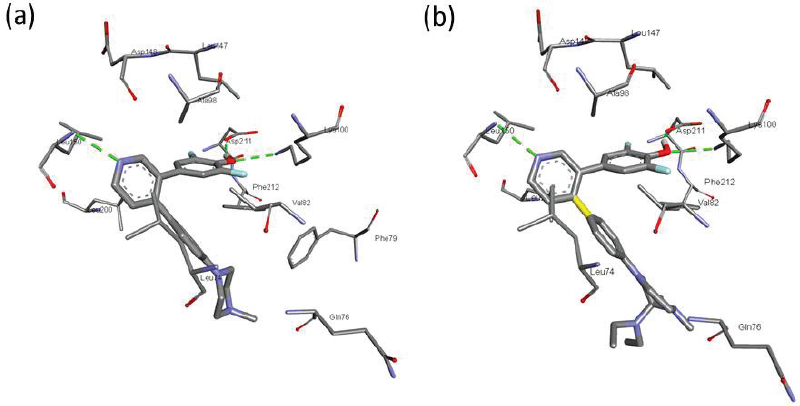 | Figure 5. The docked conformation of (a) LJH685 and (b) HA25 bound to RSK2. [Click here to view] |
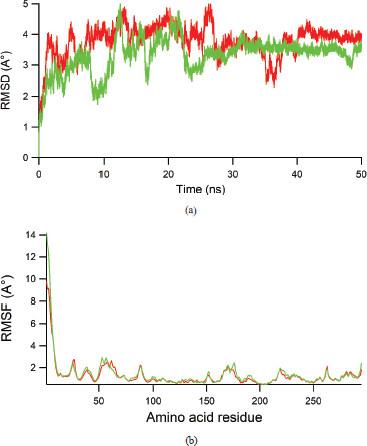 | Figure 6. (a) RMSD value of heavy atoms of RSK2 during 50 ns dynamics runs for LJH685 (red) and HA25 (green). (b) RMSF value of each amino acid residue during 50 ns dynamics simulation for LJH685 (red) and HA25 (green). [Click here to view] |
Interaction mechanism of compound
Following QSAR analysis, molecular docking was conducted to examine ligand–protein interaction. In the Surflex-Dock module, docking result was represented in Total Score, which estimates binding affinity of ligand originating from contribution of various interactions. It was found that the new candidate compound (HA25) has a higher total score (total score = 12.3519) than that of reference compound (Compound 18, total score = 9.1220).
The designing compound using 3D-QSAR CoMFA and CoMSIA models was verified through molecular docking study to confirm the binding mode with the RSK protein. As a reference, native ligand of RSK2 (PDB ID: 4NUS), i.e., LJH685, which was known as a potent RSK inhibitor, was used. LJH685 is the same compound with the most active molecule in the reference journal, which is mentioned as Compound 18 in the present study. Figure 5 shows the interaction pose of HA25 and LJH685. As shown in Figure 4, difluorophenyl ring is bound to the gatekeeper area and flipped around 49 degrees from the pyridine ring plane (Aronchik et al., 2014). The difluorophenyl ring was in hydrogen bond contact with Asp211 and Lys100, while pyridine ring was in hbond contact with Leu150. The similar interaction was found for HA25 (Fig. 4b). The hbond contacts were established between pyridine ring with Leu150, as well as between difluorophenyl and Lys100 and Asp211. The amino acid residue Phe211 was in close hydrophobic interaction with the phenyl ring of both compounds.
 | Table 4. The binding free energy terms (kcal/mol) of each LJH685 and HA25 bound to RSK2. [Click here to view] |
To assure the stability of each LJH685 and HA25 in the binding site of RSK2, MD simulation was performed for 50 ns and several dynamics properties of complex were extracted. Figure 6a shows the root mean square deviation (RMSD) for backbone atoms of RSK protein during 50 ns, while Figure 6b displays the amino acid residues fluctuation (RMSF) during 50 ns. In the RMSD graph, fluctuation was recorded in the first 25 ns, while stable dynamics were observed in the remaining time of simulation. In a similar way, the RMSF plot shows that ligand binding to the RSK2 did not induce large movement in the amino acid fluctuation. Therefore, both complexes show well stable interaction.
The last step in our study was to calculate the binding affinity of each ligand using MM-PBSA method, which is well known for its compromise between accuracy and computational cost. The method is well accepted in the calculation of ligand-receptor affinity. The MM-PBSA calculation was performed for 10,000 frames extracted from the last simulation. Table 4 shows the binding free energy and its individual component. The electrostatic energy term was lower in HA25 (ΔEELE = −29.80 ± 3.67 kcal/mol) than that in LJH685 (ΔEELE = −26.55 ± 4.79 kcal/mol). A similar trend was observed in van der Waals energy in which HA25 (ΔEVDW = −44.28 ± 3.75) was lower than that in LJH685 (ΔEVDW = −38.70 ± 4.20 kcal/mol). The non-polar contribution of desolvation energy was almost the same for both complexes (ΔEPBSUR HA25 = −4.71 ± 0.23 vs. ΔEPBSUR LJH685 = −4.13 ± 0.27 kcal/mol). However, due to the polar contribution of desolvation energy which is more positive in HA25 (ΔEPBCAL = 45.98 ± 5.12 kcal/mol) than that in LJH685 (ΔEPBCAL = 37.34 ± 5.19 kcal/mol), the total binding free energy of both complexes was almost the same (ΔEPBTOT HA25 = −32.82 ± 3.64 kcal/mol vs. ΔEPBTOT LJH685 = −32.04 ± 3.03 kcal/mol). These indicate that the designed compound is a newly potential inhibitor for RSK2.
CONCLUSION
The present study has investigated the 3D-QSAR CoMFA and CoMSIA models for difluorophenol pyridine derivatives as RSK2 inhibitor. The developed CoMFA and CoMSIA models were both validated internally and externally. Using the validated models, new compound was designed which was more potent than the parent compound (Compound 18). The newly designed compound was investigated for its interaction with RSK2 using molecular docking and MD simulation, in which the complex was stable for 50 ns. The stable trajectory of MD was then used to perform MM-PBSA calculation, in which the new compound achieved comparable binding free energy with LJH685. This indicates the potential of the new compound to be developed as a novel inhibitor of RSK2.
REFERENCES
Sulzmaier FJ, Young-Robbins S, Jiang P, Geerts D, Prechtl AM, Matter ML, Kesari S, Ramos JW. RSK2 activity mediates glioblastoma invasiveness and is a potential target for new therapeutics. Oncotarget, 2016; 7(48):79869–84. CrossRef
Stratford AL, Reipas K, Hu K, Fotovati A, Brough R, Frankum J, Takhar M, Watson P, Ashworth A, Lord CJ, Lasham A, Print CG, Dunn SE. Targeting p90 ribosomal S6 kinase eliminates tumor-initiating cells by inactivating Y-box binding protein-1 in triple-negative breast cancers. Stem Cells, 2012; 30(7):1338–48. CrossRef
Ståhle L, Wold S. Multivariate data analysis and experimental design in biomedical research. Progr Med Chem, 1988; 25:291–338. CrossRef
Smolen GA, Zhang J, Zubrowski MJ, Edelman EJ, Luo B, Yu M, Ng LW, Scherber CM, Schott BJ, Ramaswamy S, Irimia D, Root DE, Haber DA. A genome-wide RNAi screen identifies multiple RSK-dependent regulators of cell migration. Gene Dev, 2010; 24(23):2654–65. CrossRef
Romeo Y, Zhang X, Roux PP. Regulation and function of the RSK family of protein kinases. Biochem J, 2012; 441(2):553–69. CrossRef
Reipas KM, Law JH, Couto N, Islam S, Li Y, Li H, Cherkasov A, Jung K, Cheema AS, Jones SJM, Hassel JA, Dunn SE. Luteolin is a novel p90 ribosomal S6 kinase (RSK) inhibitor that suppresses Notch4 signaling by blocking the activation of Y-box binding protein-1 (YB-1). Oncotarget, 2013; 4(2):329–45. CrossRef
Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, Smith JA, Slingerland JM. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Nat Acad Sci USA, 2009; 106(23):9268–73. CrossRef
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Account Chem Res, 2000; 33(12):889–97. CrossRef
Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem, 1994; 37(24):4130–46. CrossRef
Jain R, Mathur M, Lan J, Costales A, Atallah G, Ramurthy S, Subramanian S, Setti L, Feucht P, Warne B, Doyle L, Basham S, Jefferson AB, Lindvall M, Appletin BA, Shafer CM. Discovery of potent and selective RSK inhibitors as biological probes. J Med Chem, 2015; 58(17):6766–83. CrossRef
Houles T, Roux PP. Defining the role of the RSK isoforms in cancer. Semin Cancer Bio, 2018; 48:53–61. CrossRef
Golbraikh A, Tropsha A. Beware of q2! J Mol Graph Mod, 2002; 20(4):269–76. CrossRef
Davies AH, Dunn SE. YB-1 drives preneoplastic progression: insight into opportunities for cancer prevention. Oncotarget, 2011; 2(5):401–6. CrossRef
Clark M, Cramer RD, Van Opdenbosch N. Validation of the general purpose tripos 5.2 force field. J Comput Chem, 1989; 10(8):982–1012. CrossRef
Casalvieri KA, Matheson CJ, Backos, DS, Reigan P. Selective targeting of RSK isoforms in cancer. Trends Cancer, 2017; 3(4):302–12. CrossRef
Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci, 2008; 13:4258–75. CrossRef
Bush BL, Nachbar RB. Sample-distance partial least squares: PLS optimized for many variables, with application to CoMFA. J Comput Aid Mol Des, 1993; 7(5):587–619. CrossRef
Brough R, Frankum JR, Sims D, Mackay A, Mendes-Pereira AM, Bajrami I, Costa-Cabral S, Rafiq R, Ahmad AS, Cerone MA, Natrajan R, Sharpe R, Shiu K-K, Wetterskog D, Dedes KJ, Lambros MB, Rawjee T, Linardopoulos S, Reis-Filho JS, Turner NC, Lord CJ, Ashworth A. Functional viability profiles of breast cancer. Cancer Discov, 2011; 1(3):260–73. CrossRef
Awasthi M, Singh S, Pandey VP, Dwivedi UN. CoMFA and CoMSIA-based designing of resveratrol derivatives as amyloid-beta aggregation inhibitors against Alzheimer’s disease. Med Chem Res, 2018; 27(4):1167–85. CrossRef
Aronchik I, Appleton BA, Basham SE, Crawford K, Del Rosario M, Doyle LV, Estacio WF, Lan J, Lindvall MK, Luu CA, Ornelas E, Venetsanakos E, Shafer CM, Jefferson AB. Novel potent and selective inhibitors of p90 ribosomal S6 kinase reveal the heterogeneity of RSK function in MAPK-driven cancers. Mol Cancer Res, 2014; 12(5):803–12. CrossRef
Arba M, Nur-Hidayat A, Surantaadmaja SI, Tjahjono DH. Pharmacophore-based virtual screening for identifying β5 subunit inhibitor of 20S proteasome. Comput Biol Chem, 2018b; 77:64–71. CrossRef
Arba M, Kartasasmita RE, Tjahjono DH. Molecular docking and dynamics simulations on the interaction of cationic porphyrin-anthraquinone hybrids with DNA G-quadruplexes. J Biomol Struc Dyn, 2016; 34(2):427–38. CrossRef
Arba M, Ihsan S, Tjahjono DH. Computational approach toward targeting the interaction of porphyrin derivatives with Bcl-2. J Appl Pharm Sci, 2018a; 8(12):60–6. CrossRef
Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Bio, 2008; 9:747–58.