
INTRODUCTION
The non-small cell lung cancer (NSCLC) represents the most prevalent lung cancer worldwide with less than 20% of 5-year survival rate after diagnosis (Chen et al., 2018; Gaber et al., 2018; Hao et al., 2018). The epidermal growth factor receptor (EGFR) is a member of the protein kinase family, which is a clinically validated target for NSCLC treatment. EGFR is involved in various cellular signaling cascades, which are crucial in cell growth, proliferation, survival, and migration. Due to its crucial role, there have been continuous efforts to find a small molecule that is able to inhibit EGFR, particularly for the NSCLC treatment. Erlotinib and Gefitinib were considered as the first-generation of EGFR inhibitors, which were used for the treatment of NSCLC (Bonomi, 2003; Vansteenkiste, 2004). However, it was known that the T790M point mutation in the EGFR, which was the substitution of Thr790 with Met residue, had induced acquired resistance after a median of 10–14 months to most NSCLC patients of first-generation EGFR inhibitors (Kobayashi et al., 2005). Furthermore, Afatinib and canertinib (Ou, 2012), two of several second-generation EGFR inhibitors, were approved by FDA for the treatment of metastatic NSCLC patients. However, the nonselective inhibition against wild type of EGFR has limitations in their clinical use (Gaber et al., 2018). The third-generation EGFR inhibitors such as rociletinib and avitinib (Walter et al., 2013) were developed; however, it was reported that the hyperglycemia was observed in NSCLC patients who used Rociletinib (Chen et al., 2018; Yver, 2016).
Lin et al. (2017) designed and synthesized a series of phosphoramide mustard functionality, which was incorporated into the quinazoline scaffold, and their potential as EGFR inhibitors for the treatment of lung cancer was investigated. It was found that the designed compound could inhibit EGFR with IC50 at the nanomolar range and showed no acute toxicity to mice at a single dose up to 900 mg/kg. It was concluded that the designed compound posed a potential as EGFR inhibitor. Based on these results, the present study was aimed to build a model of three-dimensional quantitative structure-activity relationship (3D-QSAR) including comparative molecular field analysis (CoMFA) (Cramer et al., 1988) and comparative molecular similarity indices analysis (CoMSIA) (Klebe et al., 1994) of quinazoline–phosphoramidate mustard conjugates. Using the built model, a novel compound was proposed, and molecular docking and molecular dynamics simulation were then used to check the conformational stability of the newly proposed compound in the binding site of EGFR.
MATERIALS AND METHODS
17 compounds of quinazoline–phosphoramidate mustard conjugates were selected based on the literature study (Lin et al., 2017). Based on a random selection, the compounds were grouped into a training set (13 compounds) and a test set (four compounds) (Table 1) by considering structural diversity and distribution of biological data. The inhibitory activity [IC50 (nM)] values were converted to the logarithmic scale (pIC50), where pIC50 = −Log IC50 (Table 2). The training set was used to develop 3D-QSAR, including CoMFA and CoMSIA, while the test set was used to evaluate the model’s validity. All structure sketching, optimization, and modeling were conducted with SYBYL-2.1 program package (Tripos, Inc.).
Minimization and alignment
Each compound was sketched and energy-minimized using Tripos molecular mechanic force field and Powell method (Clark et al., 1989), while charges were assigned using Gasteiger–Huckel method (Purcell and Singer, 1967) in the SYBYL-X 2.1. The minimization was conducted using energy convergence threshold and maximum iterations of 0.5 kcal/mol and 1,000 cycles, respectively. Superpositioning of ligands was conducted based on the core structure, N-phenylquinolin-4-amine, by employing compound 12 as a template, since it was the most active compound. Figure 1 depicts the superimposed structures of aligned molecules.
CoMFA and CoMSIA
Both CoMFA and CoMSIA were developed employing SYBYL-X 2.1 (Tripos, Inc., USA). CoMFA steric field was developed based on van der Waals interaction using Lennard-Jones potential, while CoMFA electrostatic field was based on Coulombic potential. Both CoMFA fields were generated at each lattice point of a grid box of 2.0 Å. Cut-off values of 30 kcal/mol were set for both steric and electrostatic fields (Ståhle and Wold, 1988).
In CoMSIA, a distance-dependence Gaussian-type of the physicochemical property has been adopted to avoid any singularities at the atomic position (Klebe et al., 1994). Similar standard parameters and no arbitrary cutoff limits constructed for CoMFA field calculation were used for the calculation of CoMSIA field including steric (S), electrostatic (E), hydrophobic effects (H), and hydrogen bond donor (HBD) (D) and hydrogen bond acceptor (HBA) (A).
 | Table 1. Chemical structure of quinazoline–phosphoramidate mustard conjugates and their inhibitory activities. [Click here to view] |
 | Table 2. The observed pIC50s and predicted pIC50s of the training and test set molecules. [Click here to view] |
 | Figure 1. 3D-QSAR structure superposition and alignment of the training set (left) and common substructure used for alignment (right). [Click here to view] |
The partial least squares method with leave-one-out cross-validation was employed to correlate the CoMFA electrostatic and steric fields and CoMSIA electrostatic, steric, HBD, HBA, and hydrophobic properties, each with EGFR inhibitory activity (Bush Bruce and Nachbar, 1993). The value of filtering (σ) column was set to lower than 2.0 kcal/mol to improve the signal-to-noise ratio. The optimal number of principal component (ONC) was obtained by applying LOO cross-validation, which was then utilized to derive the final CoMFA and CoMSIA models. The following equation was utilized to calculate the cross-validation coefficient of q2 value:
Where y and ŷ are observed and predicted activities of compound i, respectively, and ȳ is the average observed activity of the compound in the training set. The best QSAR model was justified on the basis of high q2, conventional correlation coefficient R2 values (q2 > 0.50 and R2 > 0.60), low standard error estimation (SEE), and an optimal number of component values.
Molecular docking
The Surflex-Dock module in SYBYL was utilized to perform molecular docking to clarify the binding mode of the compounds. The X-ray crystal structure of EGFR that was co-crystallized with Erlotinib (PDB entry code: 1M17, resolution: 2.6 Å) was taken from the RCSB Protein Data Bank. The protein structure was prepared by removing water molecules and cognate ligand and adding polar hydrogen atoms. Protomol was generated based on ligand mode, which represented a three-dimensional space in which ligand make potential interaction with every binding site.
Automatic docking was applied for molecular docking. Other parameters were left at default. Total-score of Surflex-Dock, which was expressed in the negative logarithm of the dissociation constant, –log10 (Kd), was used to represent binding affinities. The docked conformation of the ligand was generated after docking, where those with the highest scores were selected as the docking results. Each compound was energetically minimized using the Tripos force field and the Powell algorithm with a convergence criterion of 0.05 kcal/mol A° and Gasteiger–Huckel charges.
Molecular dynamics simulation and MM-PBSA calculation
Molecular dynamics (MD) simulation of 100 ns was performed for Compound 12 (Comp12), RA1, RA2, RA3, and RA4, each complexed with EGFR. The details of MD protocol follow our previous work (Arba et al., 2018a). Molecular mechanics-Poisson Boltzmann Surface Area (MM-PBSA) (Kollman et al., 2000) calculation was performed to predict the binding free energy of Comp12, RA1, RA2, RA3 and RA4 to EGFR. The MM-PBSA calculation was performed using 1,500 snapshots taken from 85 to 100 ns. Details of MM-PBSA protocol were explained in Kollman et al. (2000), Arba et al. (2018b) and Arba et al. (2018c).
RESULTS AND DISCUSSION
CoMFA model
The quinazoline–phosphoramidate mustard conjugates were utilized to conduct the CoMFA study. The CoMFA model calculated from the training set exhibited good cross-validated correlation coefficient with q2 = 0.577, r2 = 0.982, F = 166.591, and SEE = 0.09, with three ONC. The external validation of the CoMFA model using the test set showed good predicted correlation coefficient r2pred = 0.926, which indicated the predictive ability of the model. Table 2 shows the actual and predicted pIC50 as well as the residuals of the training and test set compounds, while Table 3 shows the statistical parameters associated with the CoMFA model. The steric and electrostatic field contributions were found to be 37.4:62.6, which indicated the significant contributions of both fields on ligand-receptor interaction.
CoMSIA model
Compared to CoMFA, CoMSIA defines five interaction fields, i.e., steric, electrostatic, hydrophobic, HBD, and HBA. The final CoMSIA model gave cross-validated correlation coefficient q2 = 0.537, r2 =0.964, and SEE=0.13 for three numbers of component. The external validation of the test set resulted in r2pred = 0.921, which indicated the predictive ability of the CoMSIA model. The field contribution values for steric, electrostatic, hydrophobic, HBD, and HBA were 8.6%, 31.7%, 20.7%, 21.2%, and 17.8%, respectively. Figure 2 exhibits the relationship between the data of predicted and observed activity for CoMFA and CoMSIA models.
 | Table 3. PLS statistics of CoMFA and CoMSIA models. [Click here to view] |
 | Figure 2. Scatter plot diagram between actual pIC50 versus predicted pIC50 for (a) CoMFA and (b) CoMSIA. [Click here to view] |
Graphical interpretation of CoMFA and CoMSIA
CoMFA and CoMSIA contour maps were generated to rationalize the regions in 3D space around the molecules for increasing the inhibitory activity. The CoMFA steric and electrostatic contour maps are shown in Figure 3, while the corresponding CoMSIA contour maps are shown in Figure 4. The most active compound 12 was used as the reference structure.
CoMFA contour maps
CoMFA steric interactions are represented by green and yellow colored contours, while CoMFA electrostatic interactions are shown with red and blue colored contours. The bulky substituents are favorable in the green regions of steric contours for enhancing the inhibitory activity, while those in yellow regions may lead to a decrease in inhibitory activity. Meanwhile, in the map of the electrostatic field, the blue contours indicate that electropositive charges are favored for inhibitory activity, while the red contour designates an increase in inhibitory activity of the electronegative charges. Compound 12 (Comp12) was utilized to explain the contour map. Figure 3 exhibits CoMFA steric and electrostatic contour maps. The steric field contour shows that the small-sized green contour could be observed near R1 position, which indicates that the addition of small bulky groups near those green regions would increase the inhibitory activity. On the other hand, the yellow contour around the R2 position indicates that hydrophobic or bulky group substitution near the yellow regions is favored for increasing the activity. The CoMFA steric interaction agrees well with the experimental results. For instance, compound 11 {R1 = H, R2 = 3-Cl-4-[(3-F-benzyl)oxo]phenyl, n = 3, X = O}, compound 8 {R1 = H, R2 = 3-Cl-4-[(3-F-benzyl)oxo]phenyl, n = 4, X = O}, compound 17 {R1 = H, R2 = 3-Cl-4-[(3-F-benzyl)oxo]phenyl, n = 3, X = CONH}, and compound 14 {R1 = OMe, R2 = 3-Cl-4-[(3-F-benzyl)oxo]phenyl, n = 3, X = O} had the lowest activities as indicated by pIC50 values of 7.1249, 7.5686, 7.585, and 7.6576, respectively. On the other hand, only red contour was observed in the electrostatic contour map near R1 substituent, which indicates that electronegative substituent at the position would increase the inhibitory activity. This phenomenon explains the reason why compound 14 (R1 = OMe) displayed low inhibitory activity (IC50 = 22 nM).
CoMSIA contour map
The COMSIA steric field contour maps are depicted in Figure 4a. The green contour around R1 means that sterically, bulky groups are favorable for increasing the inhibitory activity. In contrast, a yellow contour near the phenyl group means that bulky group substituent in that position may decrease the inhibitory activity.
In the CoMSIA electrostatic field (Fig. 4b), the blue color designates the positively charged groups that are favored for inhibitory activity, while the red contour denotes the negatively charged groups, which are favored for improving the inhibitory activity. CoMSIA hydrophobic contour map is shown in Figure 4c. The hydrophobic yellow contours can be observed around the C3 position of R2, which indicates that replacing this position with hydrophobic groups may increase the activity. In contrast, the white contour around the C4 position of R2 indicates that hydrophobic groups are not favored for increasing the activity.
 | Figure 3. CoMFA steric (a) and electrostatic (b) contour maps with 2 Å grid spacing. Compound 12 was displayed in the background. In steric and electrostatic fields, the contribution of green and blue contours, respectively, is 80%, while those of yellow and red, respectively, is 20%. [Click here to view] |
 | Figure 4. CoMSIA std*coeff contour maps with compound 12 as a background. (a) Steric fields: contribution of green contours is 80%, while yellow contours are 20% contribution. (b) Electrostatic fields: blue contours are 80% contribution, while red contours are 20% contribution. (c) Hydrophobic fields: yellow contours are 80% contribution, while white contours are 20% contribution. [Click here to view] |
In the CoMSIA HBA (Fig. 5a), the magenta contour represents that the hydrogen bond acceptor groups are favorable for increasing the inhibitory activity, while the red contour implies that the HBA groups would decrease the inhibitory activity. In the CoMSIA HBD (Fig. 5b), HBD groups are favored in the cyan contour for increasing the inhibitory activity, while the purple contour denotes that HBD groups are not preferred. Overall, electrostatic field contributes the most to the CoMSIA model as shown in Table 3, which indicates that the electrostatic field is the most influencing factor to the inhibitory activity of the quinazoline–phosphoramidate mustard conjugates. This result agrees well with the CoMFA model, which shows that the electrostatic field is more important to the inhibitory activity than the steric field (Table 3).
Design for new compound
Based on the proposed 3D CoMFA and CoMSIA models, new compounds are designed using compound 12 (Comp12) as a template. All new compounds were minimized and aligned to the database, and then docked into the active site of EGFR. The binding affinities (total score) of the newly designed compounds were higher than that of Comp12. Table 4 depicts the structures of the newly designed compound using the predicted pIC50 CoMFA and CoMSIA as well as total scores, while Figure 6 shows predicted pIC50 values between CoMFA and CoMSIA models.
Molecular docking
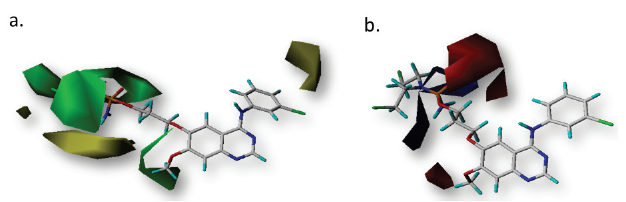
To explore the interaction of Comp12, RA1, RA2, RA3, and RA4 with EGFR, molecular docking using surflex-dock was applied. The results showed that all Comp12, RA1, RA2, RA3, and RA4 formed hbond interactions with Met769. In the crystallographic pose of erlotinib, Met769 was observed in direct hbond. Additional hbond with Thr766 was established with Comp12, RA1, and RA3, which was detected in the crystal structure of erlotinib through water-mediated hbond. Besides, RA1 and RA2 formed hbonds with Lys721, while Comp12 and RA1 share the same hbond with Thr830. Figure 7 depicts the docked positions of Comp12, RA1, RA2, RA3, and RA4 in the binding site of EGFR.
MD simulation and MM-PBSA calculation
MD simulation of 100 ns was performed on each complex of Comp12, RA1, RA2, RA3, and RA4, with EGFR. Figure 8a shows the RMSD of the heavy atoms of EGFR during 100 ns. It shows stable conformations of both complexes after 10 ns. To assess the flexibility of amino acid residues during 100 ns, RMSF plot was measured (Fig. 8b). The results showed that the two complexes had a similar pattern of residual movements. High flexibility was observed in the amino and carboxy terminals of the protein, while the majority of the residues showed rigid flexibility, which indicated that the ligand binding did not induce a large change in protein conformation.
 | Figure 5. CoMSIA std*coeff. contour maps with compound 12 as a background. (a) H-bond acceptor fields: the magenta contour is 80% contribution, while the red contour is 20% contribution. (b) H-bond donor fields: the cyan contour is 80% contribution, while purple is 20% contribution. [Click here to view] |
 | Table 4. Chemical structures of newly designed compounds and their predicted pIC50 CoMFA and CoMSIA models as well as total scores. [Click here to view] |
To assess the affinity of each compound to EGFR, MM-PBSA calculation was performed (Table 5). It can be inferred from Table 5 that electrostatic, van der Waals, and non-polar energy of desolvation were favorable for ligand binding in both complexes. Whereas, the polar energy of desolvation was not favored, resulting in unfavorable net electrostatic energies. Interestingly, the predicted total binding free energy of RA2 (ΔG = −38.32 kcal/mol) was lower than that of Comp12 (ΔG = −30.48 kcal/mol), which indicated the good potential of the designed compound.
 | Figure 6. The pIC50 values between CoMFA and CoMSIA models. [Click here to view] |
 | Figure 7. The docked poses of (a) Comp12, (b) RA1, (c) RA2, (d) RA3, and (e) RA4. [Click here to view] |
 | Figure 8. (a) RMSD value of each ligand-EGFR complex during 100 ns dynamics runs calculated from heavy atoms of protein for Comp12 (red), RA1 (yellow), RA2 (green), RA3 (blue), and RA4 (purple). (b) RMSF plot of each amino acid residue during 100 ns dynamics simulation for Comp12 (red), RA1 (yellow), RA2 (green), RA3 (blue), and RA4 (purple). [Click here to view] |
 | Table 5. The binding free energy terms (kcal/mol) of each ligand bound to EGFR. [Click here to view] |
CONCLUSION
In the current study, CoMFA and CoMSIA analysis and molecular docking were performed to explore the structure-activity relationship of novel quinazoline–phosphoramidate mustard conjugates as an EGFR inhibitor. Both CoMFA and CoMSIA models were valid with acceptable statistical criteria. Using CoMFA and CoMSIA contour maps, new compounds were designed and docked to the binding site of EGFR. The docked position of the newly designed compounds showed key interactions with the active site residues of EGFR, which was stable during 100 ns of MD simulation. The MM-PBSA binding energy analysis shows that one of the new compounds had higher affinity than that of the parent compound, thus providing a good candidate for further drug discovery research.
ACKNOWLEDGMENTS
The authors wish to thank Universitas Halu Oleo, Ministry of Research, Technology and Higher Education Republic of Indonesia for supporting this research through Hibah Penelitian Dasar 2018.
REFERENCES
Arba M, Nur-Hidayat A, Ruslin, Yusuf M, Sumarlin, Hertadi R, Wahyudi ST, Surantaadmaja SI, Tjahjono DH. Molecular modeling on porphyrin derivatives as β5 subunit inhibitor of 20S proteasome. Comput Biol Chem, 2018a; 74:230–8.
Arba M, Nur-Hidayat A, Surantaadmaja SI, Tjahjono DH. Pharmacophore-based virtual screening for identifying β5 subunit inhibitor of 20S proteasome. Comput Biol Chem, 2018b; 77:64–71.
Arba M, Ruslin, Kalsum WU, Alroem A, Muzakkar MZ, Usman I, Tjahjono DH. QSAR, molecular docking and dynamics studies of quinazoline derivatives as inhibitor of phosphatidylinositol 3-kinase. J Appl Pharm Sci, 2018c; 8(05):001–9.
Bonomi, P. Erlotinib: a new therapeutic approach for non-small cell lung cancer. Expert Opin Inv Drug, 2003; 12(8):1395–401.
Bush Bruce L, Nachbar RB. Sample-distance partial least squares: PLS optimized for many variables, with application to CoMFA. J Comput Aid Mol Des, 1993; 7(5):587–619.
Chen L, Fu W, Zheng L, Liu Z, Liang G. Recent progress of small-molecule epidermal growth factor receptor (EGFR) inhibitors against C797S resistance in non-small-cell lung cancer. J Med Chem, 2018; 61(10):4290–300.
Clark M, Cramer RD, Van Opdenbosch N. Validation of the general purpose tripos 5.2 force field. J Comput Chem, 2018; 10(8):982–1012.
Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc, 1988; 110(18):5959–67.
Gaber AA, Bayoumi AH, El-morsy AM, Sherbiny FF, Mehany ABM, Eissa IH. Design, synthesis and anticancer evaluation of 1H-pyrazolo[3,4-d]pyrimidine derivatives as potent EGFRWT and EGFRT790M inhibitors and apoptosis inducers. Bioorg Chem, 2018; 80:375–95.
Hao Y, Lyu J, Qu R, Tong Y, Sun D, Feng F, Tong L, Yang T, Zhao Z, Zhu L, Ding J, Xu Y, Xie H, Li H. Design, Synthesis, and biological evaluation of pyrimido[4,5- d ]pyrimidine-2,4(1 h ,3 h )-diones as potent and selective epidermal growth factor receptor (EGFR) inhibitors against L858R/T790M resistance mutation. J Med Chem, 2018; 61(13):5609–22.
Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem, 1994; 37(24):4130–4146.
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non–small-cell lung cancer to Gefitinib. New Engl J Med, 2005; 352(8):786–92.
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Account Chem Res, 2000; 33(12):889–97.
Lin S, Li Y, Zheng Y, Luo L, Sun Q, Ge Z, Cheng T, Li R. Design, synthesis and biological evaluation of quinazoline–phosphoramidate mustard conjugates as anticancer drugs. Eur J Med Chem, 2017; 127:442–58.
Ou, SHI. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? A review of the clinical evidence. Crit Rev Oncol Hemat, 2012; 83(3):407–21.
Purcell WP, Singer JA. A brief review and table of semiempirical parameters used in the Hueckel molecular orbital method. J Chem Eng Data, 1967; 12(2):235–46.
Ståhle L, Wold S. Multivariate data analysis and experimental design in biomedical research. Progr Med Chem, 1988; 25:291−338.
Vansteenkiste JF. Gefitinib (Iressa®): a novel treatment for non-small cell lung cancer. Expert Rev Anticanc, 2004; 4(1):5–17.
Walter AO, Sjin RTT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, Martin TS, Karp R, Kalken DV, Chatuverdi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Dyke TV, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC, Allen A. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M mediated resistance in NSCLC. Cancer Discov, 2013; 3(12):1404–15.
Yver A. Osimertinib (AZD9291)-a science-driven, collaborative approach to rapid drug design and development. Ann Oncol, 2016; 27(6):1165–70.