
INTRODUCTION
Hypertension is one of the foremost manageable risk factors for many cardiovascular diseases. Elevated blood pressure has become one of the major contributors to global mortality (Zeng et al., 2020). Hypertension can be controlled in cardiovascular disease patients. Uncontrolled hypertension accounts for about 45% of global cardiovascular diseases’ morbidity and mortality (Costa et al., 2018). Hypertension is a prevalent disease all over the world. The global prevalence of hypertension has greatly increased, where one in every four men and one in every five women have hypertension (Zeng et al., 2020). At the beginning of the 20th century, almost 972 million people over the world had hypertension. It is predicted that over 1.56 billion people worldwide will be suffering from hypertension by 2025 (Zhang et al., 2017).
Thus, it is a requisite to alleviate high blood pressure, to avoid its consequent threats. Agents blocking the renin-angiotensin system (RAS), known as RAS-acting agents, are considered as a promising and effective approach for the treatment of hypertension and congestive heart failure. RAS-acting agents are recommended for the first-line treatment of hypertension, as they have been proved to reduce mortality and morbidity (El-Gendy et al., 2017). One of the most common medications used in the treatment of hypertension is angiotensin II receptor blockers (ARBs), as they are nonpeptide angiotensin II receptor antagonists that block the angiotensin II type receptor. This group was developed to minimize side effects observed by the angiotensin-converting enzyme inhibitors through fully blocking RAS (Brunner, 2007, Kearney et al., 2003).
The antihypertensive drug olmesartan medoxomil (OLM), which belongs to the ARBs family (Li et al., 2016), was chosen as a drug model in this study because of its prospective benefits. The once-daily dosing, absence of significant side effects, and fewer adverse effects than the other ARBs family members, valsartan, losartan, and amlodipine, contribute to the OLM benefits (Zhang et al., 2017). According to the Biopharmaceutics Classification System (BCS), it is classified as a class II drug which is practically insoluble in water, resulting in its poor oral bioavailability (26%). These bioavailability limitations lead to the rejection of almost 40% of invaluable active ingredients and impede their formation in the development stage. Moreover, about 70% of the new chemical entities that enter the drug development stage have been estimated to have insufficient solubility in water and hence low absorption from the gastrointestinal tract (El-Gendy et al., 2017). The increased number of highly lipophilic compounds increased the interest to deliver these poorly soluble drugs through alternative routes, other than the per-oral route (El-Setouhy et al., 2015). Nowadays, orally disintegrating tablets (ODTs) have become increasingly popular and welcome, due to their ease of self-administration, chiefly in pediatrics and geriatrics, in addition to their suitability to be used in therapy modules of dysphagia and also for patients who have difficulty in movement, as well as patients with limited or no access to water (Gugulothu et al., 2015). According to the United States Food and Drug Administration, ODTs are solid dosage forms comprising active pharmaceutical ingredients which break down rapidly, usually within a few seconds, when placed upon the tongue (Abay and Ugurlu, 2015).
There are numerous techniques applied for preparing ODTs like the addition of superdisintegrants using direct compression (Singh et al., 2009), spray drying (Mishra et al., 2006), sublimation (Narmada et al., 2009), mass extrusion, tablet molding (Gugulothu et al., 2015), lyophilization (Safar et al., 2011), and others (Chauhan et al., 2018). As an increasing number of product approvals have been provided in the past few years, lyophilization has been considered as the most advantageous and cost-effective approach to developing orodispersible lyophilisates (ODLs) formulated for large-scale production (Dey and Ghosh, 2016; Kassem and Labib, 2016). ODLs disintegrate or dissolve in the oral cavity in a very short time (the shortest among other technologies), owing to their uniform, highly porous structure, high specific surface area, and hydrophilic matrix (Fouad et al., 2020; Gugulothu et al., 2015; Kassem and Labib, 2016; Safar et al., 2011). Lyophilization (freeze-drying) is the process in which the solvent is sublimed from a frozen solution or suspension with a structure-forming additive. The entire freeze-drying process is carried out at nonraised temperatures to avoid adverse thermal effects that may affect the active ingredient’s stability during drying (Kathpalia et al., 2013; Liew et al., 2016). A further credit to the lyophilization process is that it creates a glassy amorphous structure of the drug together with the additives (Fu et al., 2004). The amorphous structure can dissolve instantly upon contact with saliva leading to enhancement of the dissolution rate of the drug and ultimately reveal improved absorption and bioavailability (Chauhan et al., 2018; Kassem and Labib, 2016).
A comprehensive understanding of the process and the material parameters is a successful way to control the quality attribute. The usual empirical screening methods are time-consuming and do not give the whole effects of the process and formulation factors (Iurian et al., 2017). Thus, a deep understanding and control of the whole pharmaceutical development process became a crucial step, to ensure process and product robustness. The full understanding of the product and the process could be achieved by applying quality by the design (QbD) concepts, which was defined by the ICH Q8 as “a risk-based assessment method, with a predefined objective, comprehensive understanding of the process and product that ensures building of the quality into the product during its design rather than being tested” (Xu et al., 2012). Application of this concept, process, and product variables could be well understood, with the creation of a design space, to ensure the fulfillment of the objectives and characteristics (Mishra and Rohera, 2017). Hence, the current study aims to develop and optimize ODLs containing OLM, to enhance its solubility and bioavailability, to give a fast onset of action, by applying QbD concepts.
MATERIALS AND METHODS
Materials
OLM was kindly supplied by Rameda Pharmaceutical Company, Giza, Egypt. Soluplus® was obtained from BASF SE, Ludwigshafen, Germany. Mannitol was kindly provided by Roquette Pharma (Lestrem, France). Gelatin, glycine, sodium alginate, and sucralose were obtained from El-Nasr Pharmaceutical Chemicals Co., Cairo, Egypt. Hydroxypropyl methylcellulose (HPMC), Na-carboxymethyl cellulose (Na-CMC), and croscarmellose sodium were purchased from Adwic Pharmaceutical Company, Cairo, Egypt. F-melt and PEARLITOL Flash were purchased from Fuji Chemical Industry Co., Ltd., Toyama, Japan. Valsartan, β-cyclodextrin, polyvinyl pyrrolidone (PVP K90) and Brij S20, acetonitrile [ACN, ≥99.9%, High performance liquid chromatography (HPLC) grade], ammonium acetate [Liquid chromatography [liquid chromatography (LC)/mass eluent additive], hydrochloric acid, and tertiary butyl methyl ether (>99.8%, high performance liquid chromatography (HPLC) grade) were purchased from Sigma-Aldrich (St. Louis, MO).
Preparation of OLM-ODLs
Different OLM-ODLs were developed using different polymers. These polymers contribute with many roles in the ODL formulations, such as being a diluent, binder, solubility enhancer for poorly water-soluble drugs, and superdisintegrant, which provide short disintegration time. Glycine is another additive used. It is one of the amino acids which exhibit an excellent wetting property and is highly recommended to be used in fast-disintegrating tablets manufacturing. The addition of glycine is proved to increase the stability of the formulation (Fukami et al., 2006). Mannitol was used as a bulking agent and is also useful for decreasing the friability and increasing the hardness of the formulated tablets. A range of semisynthetic superdisintegrants were used to promote the drug release by enhancing water wicking into the tablet. Furthermore, they promote the deaggregation of the tablet particles. Sucralose is an artificial sweetener, which is widely preferable to be used in pharmaceutical formulations, especially oral disintegration tablets. It shows some benefits, such as increasing formulation stability due to its pH and heat stability. Moreover, it shows rapid solubility, besides its ease of handling, being inert, and not interacting with the formulation ingredients. Furthermore, it is suitable for pregnant women, children, and glucose-intolerant and diabetic patients (Al Humaid, 2018).
OLM-ODLs were prepared using the freeze-drying technique in an aqueous solution including the matrix former and other additives. An exactly weighed amount of OLM powder was dispersed using a magnetic stirrer (MMS-3000, Biosan Ltd., UK) to yield a concentration of 40 mg/ml. The final suspension was distributed into round-shaped blisters and frozen overnight at −20°C and thereafter lyophilized for 24 hours under a pressure of 7 × 10−2 mbar at −45°C using a Novalyphe-NL 500 freeze-dryer (Savant Instruments Corp., USA) (Basha et al., 2020).
Furthermore, different formulae were also prepared in the presence of four different solubilizers in three different ratios to the earlier mentioned aqueous suspension, namely, Soluplus, β-cyclodextrin, PVP K90, and Brij S20. It should be noted that the higher and lower levels of the used solubilizers were used based on previous literature and preliminary experiments (Hirlekar et al., 2009; Shamma and Basha, 2013; Volkova et al., 2021), whereas the β-cyclodextrin was used at a ratio between it and the drug of 1:1, 1:2, or 1:3, not as percentages like the rest of the solubilizers (Abdel Halim, 2013). The ODLs were stored in securely closed containers in desiccators over anhydrous calcium chloride at 25°C until needed for extra investigations (Abdel Halim, 2013).
QbD paradigm
Quality target product profile (QTPP) and risk identification
QTPP could be defined according to the ICH Q8 (R2) as “a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product” (ICH Expert Working Group, 2009). In this study, the QTPP was to enhance the solubility and bioavailability of OLM, a BCS class II, to achieve fast drug release. Accordingly, preformulation studies and data from the literature review were collected to create the knowledge space of the study. According to the US FDA, in 2008 guidance about oral disintegrating tablets formulations, the ODTs should disintegrate within 30 seconds or less. In addition, ODTs should show an appropriate degree of friability, less than 1%, to withstand packaging and transportation. Besides, fast drug release is expected from ODTs, to achieve the required target (Mishra and Rohera, 2017). Thus, the most influential parameters affecting the ODTs could be considered as tablet friability, wetting and disintegration time, and the cumulative amount of drug released within 15 minutes (Q15%), which are taken to be the critical quality attributes (CQAs) in this study. The whole steps in the QbD study were schematically presented in Figure 1.
Risk analysis (RA)
Checking the possible hazards, and analyzing the factors affecting the CQAs, starts with a detailed risk assessment study. The first step was the identification of the CQAs, which was followed by the determination of the critical process parameters (CPPs) and material attributes (MAs), which may have a direct impact on the QTPP. This step was hierarchically represented using an Ishikawa diagram, as represented in Figure 2. Ishikawa diagrams are used to potentially study the whole factors affecting the CQAs. Figure 2 shows how the CQAs are affected by each of the methods, material, equipment, and environment, which all helped greatly to identify the failure modes of the ODLs formulations (Amasya et al., 2016). As a result, the effects of the friability %, wetting time, disintegration time, and drug release rate were found to be the most CPPs and MAs affecting the CQAs.
Screening of matrix formers and superdisintegrants on the quality characteristics of ODLs
A crucial and very important step in selecting the CPPs and MAs which significantly affects the CQAs is the screening step (Singh et al., 2018). As the main aim of the current study is to prepare ODTs to facilitate fast absorption, the choice of the matrix former type was very important. Accordingly, the first step was the screening of the effect of the type of the matrix former on the ODLs. The effect of the matrix former on the properties of the ODL was studied by preparing S1–S4, as tabulated in Table 1, containing four different types of matrix formers, namely, sodium alginate, HPMC, sodium CMC, and gelatin. The matrix former type was selected based on the previous literature (Abd Elbary et al., 2012; Shoukri et al., 2009). The superdisintegrant used was croscarmellose, where the drug content, friability, cumulative amount of drug released within 15 minutes (Q15%), disintegration time, and wetting time were tested (Shoukri et al., 2009).
After screening the effect of the matrix former type, and selection of the best based on the characterization test, the effect of the superdisintegrant type on the ODLs was screened, owing to the great importance of the superdisintegrants in the ODL formulation (Tashan et al., 2020). Thus, the best matrix former formula was prepared with one of another two superdisintegrants, F-melt and PEARLITOL Flash (S5-S6), and was evaluated in terms of the aforementioned tests. The whole screening design is tabulated in Table 1.
 | Table 1. Formulations content, and their results for the screening step. [Click here to view] |
Optimization of OLM-ODLs using D-optimal design
The screening step resulted in choosing the best matrix former and the best superdisintegrant for the optimization step, which were the gelatin and the supercarmellose, respectively. Additionally, a solubilizer was added to enhance the disintegration of the ODL. A D-optimal design was the design of choice for the optimization of OLM-ODLs. The design was employed to evaluate the effect of each of the solubilizer types (X1) (Soluplus, β-cyclodextrin, PVP K90, and Brij S20) and the concentration of the solubilizer (1%–3%) (X2). The effect of the solubilizer type was studied due to its crucial role in improving the solubility of poorly water-soluble drugs, thus enhancing their biopharmaceutical performance (Shamma and Basha, 2013). This resulted in the formulation of 19 formulae as represented in Table 2, using Design-Expert® 10.0.1.0 software (Stat-Ease Inc., USA). These 19 formulae were tested in terms of disintegration time (Y1) and cumulative amount of drug released at 15 minutes (Q15%) (Y2), as they may have a greater effect on the QTPP of the current study. A second-order polynomial model was constructed.
Furthermore, numerical optimization was applied based on the desirability approach, resulting in an optimized formula with the desired CQAs. The optimized formula, as suggested by the software, was prepared, evaluated in terms of the previously mentioned CQAs, and compared with the expected results to obtain the % bias, thus checking the validity of the design (Sweed et al., 2021).
Evaluation of OLM-ODLs
Uniformity of ODL thickness and diameter
The integrity, shape, and overall elegancy of ODLs are major parameters for patient compliance. Therefore, attributes concerning the general appearance of the ODLs’ color, shape, surface texture, and consistency were evaluated visually. Samples of 10 tablets of the optimized formula were checked out for thickness and diameter by using a digitalized vernier caliper (Mitutoyo, Tokyo, Japan), and the mean value was calculated (Basha et al., 2020).
 | Table 2. Levels of the MA in the D-optimal design, formulations and their results. [Click here to view] |
Weight uniformity
The weight variation test was conducted by weighing 10 ODLs of the optimized formula separately, and, afterward, the average weight of the ODLs was measured and compared to the individual tablet weights. The percentage of weight variation was mathematically calculated according to the following equation (Basha et al., 2020):
Drug content uniformity
Ten ODLs from each batch were used in this test, where each tablet was crushed and dispersed in a 50 ml volumetric flask containing 10 ml ethanol and sonicated (Model 2210, Branson Ultrasonics Co., Danbury, CT) for 30 minutes to allow complete dissolution. Then, the solution was completed to 50 ml with phosphate buffer saline at pH 6.8 and filtered using a membrane filter (0.45 μm), suitably diluted, where the OLMs content was analyzed spectrophotometrically at predetermined maximum wavelength λmax 257 nm (Shimadzu UV spectrophotometer, 2401/PC, Japan) (Gugulothu et al., 2015).
In vitro disintegration time
In vitro disintegration time for ODLs was calculated using a United Stated Pharmacopeia (USP) disintegration tester (Logan Instruments Corp., NJ), where 500 ml of distilled water was used as the disintegration medium, at 37°C ± 0.5°C. Six ODLs of each formula were allocated into a basket rack and covered with a transparent plastic disc. The time taken for complete disintegration of the ODLs, after passing them through the screen of the basket rack with no observable mass remaining in the apparatus, was determined in seconds (Gugulothu et al., 2015; Shoukri et al., 2009).
Friability test
Twenty pre-weighed ODLs of each type of the prepared formulations were tested for friability, to evaluate the effect of friction and shocks, which may cause ODLs to flake, cap, or crack. A friability apparatus (ERWEKA-GmbH, Germany) was used for this purpose. The drum was rotated at 25 rpm for 4 minutes. Then ODLs were taken out, dedusted, and reweighed. The percentage friability was calculated from the loss in weight as given in the following equation (Shoukri et al., 2009):
Wetting time test
A piece of filter paper was placed in a Petri dish of a 10 cm diameter. Distilled water (10 ml) containing 2% w/v methylene blue, a water-soluble dye, was added to the Petri dish. One ODL of each type was carefully put down on the surface of the filter paper, and the time required for colored water to reach the ODL upper surface was recorded using a stopwatch as the wetting time. The blue dye solution was used to enable suitable visualization (Abd Elbary et al., 2012). The test was carried out in triplicate.
In vitro drug release
The dissolution profiles of OLM from different formulations compared with the plain drug were determined, using a USP rotating paddle apparatus (Hanson SR8-Plus, USA), rotating at 75 rpm and maintained at 37°C ± 0.5°C. All tests were conducted in 250 ml phosphate buffer saline (pH = 6.8) as a dissolution medium. The amount of drug used was equivalent to 20 mg. Samples of 5 ml were withdrawn and replaced with an equal volume of fresh medium to maintain a constant total volume at specified time intervals (1, 2, 3, 5, 7, 10, 12, and 15 minutes). Samples were filtered through a 0.45 μm membrane filter, and the dissolution medium was suitably diluted, before being assayed spectrophotometrically for drug content at λmax 257 nm (Shimadzu UV spectrophotometer, 2401/PC, Japan). The cumulative amount of OLM dissolved in the formulations was calculated and plotted (expressed as a percentage of the labeled amount) as a function of time to produce the dissolution profile (Shoukri et al., 2009). All experiments were done in triplicate.
Physicopharmaceutical characterization of the optimized ODLs
Scanning electron microscope (SEM) analysis
The morphology of the optimized OLM-ODL formula was examined, by SEM analysis (JEOL-JSM-5300 SEM, Tokyo, Japan). Both surface characteristics and cross-section were investigated. Cross-section samples were prepared by cutting a thin slice of the tablet with a razor blade to expose the inner structure. The samples were fixed on a brass stub and then made electrically conductive by coating, in a vacuum, with a thin layer of gold (~150 Å) for 30 seconds. The pictures were taken at an excitation voltage of 20 kV (Basha et al., 2020).
Fourier transform infrared (FTIR) spectroscopy
FTIR spectra of pure OLM, pure sucralose, and the selected OLM-ODL formula were recorded using an FTIR spectrophotometer (Perkin Elmer, Germany) at a range from 400 to 4,000 cm−1 and a resolution of 4 cm−1 (Abd Elbary et al., 2012).
In vivo study
Experimental animals
The animals used in the current study were New Zealand male rabbits, after approval of MSA University Ethics Committee (Pt17/Ec16/2019 F) (Cairo, Egypt), where ARRIVAL guidelines were applied. The important rationale for using a rabbit model is that the rabbit sublingual cavity is similar to that of a human as it is a nonkeratinized mucosa which is suitable for correlating bioavailability of optimized OLM-ODL formulation with conditions persisting in human beings. In general, rabbits are considered as appropriate animal models due to the histological similarity of the oral cavity to that of humans (Aghera et al., 2012; Rawas-Qalaji et al., 2006). Twelve New Zealand male rabbits of 3 ± 0.5 kg were provided by the Animal House of MSA University (Cairo, Egypt). The rabbits were housed in stainless steel cages under light- and temperature-controlled conditions (24°C ± 2°C under a 12 hour light/dark cycle) with a 40%–60% relative humidity. The animals were provided with free access to standard laboratory food and water.
Study design
The study was performed to compare the pharmacokinetics of OLM from the optimized OLM-ODL, with OLM per-oral suspension, after the administration of single doses equivalent to 0.25 mg/kg. The first group (six rabbits) received per-oral OLM suspension, while the second group (six rabbits) received the optimized OLM-ODL formulation; ODLs were inserted sublingually. To avoid the swallowing of the ODLs by the rabbits, they were kept under anesthesia throughout the experiment. The rabbits were housed in a wooden holder so that blood samples could be taken. A 24-gauge needle was used to take blood samples from the left marginal ear vein. Following per-oral OLM suspension and optimized OLM-ODL formulation administration, 1 ml of blood was withdrawn at time points of 0 (pre-dose), 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 hours. The blood samples were collected in an ethylenediaminetetraacetic acid vacutainer and centrifuged at 3,500 rpm for 10 minutes. Plasma was transferred instantly into plastic tubes and stored frozen at −20°C for further analysis (Kumar et al., 2016).
Sample preparation
All frozen rabbits’ plasma samples were thawed at ambient temperature. Rabbit plasma samples (0.5 ml) were placed in 7 ml glass tubes, and 100 µl of internal standard solution (Valsartan), 100 µl HCl, and 5 ml tertiary butyl methyl ether were added to each. The tubes were then centrifuged at 3,000 rpm for 10 minutes. After that, the upper organic phases were moved to a clean glass tube, filtered through a 0.45 µm Millipore filter, and dried under vacuum, and the residue was reconstituted with a 25 µl mobile phase and injected into an Liquid chromatography–mass spectrometry (LC/MS) system (AB Sciex Instruments, 1007441-O). A series of standard solutions of OLM containing 6, 12, 18, 50, 250, 500, 750, 1,006, 1,404, 1,610, and 2,000 ng/ml were prepared by diluting the stock solution of the drug with the mobile phase. The samples area ratio was measured, and a calibration curve for the drug was constructed where the peak area ratio was plotted as a function of the concentration of the drug (ng/ml). The equation of the straight line was used to calculate the concentration of the drug in the plasma samples (Musijowski et al., 2015).
A triple-quadrupole LC/MS (AB Sciex Instruments, 1007441-O) was used for the estimation of the reconstituted drug in the mobile phase. The LC/MS method is a sensitive and accurate method for studying the pharmacokinetics of OLM in vivo, especially when detected at very low concentrations (Danafar and Hamidi, 2016). LC/MS analysis was performed on an Agilent-C18 column [150 × 4.6 mm, 5 μm particle size i.d. (Agilent Technologies, Palo Alto, CA)] using ACN and ammonium acetate 0.02 M (80:20 v/v) as a mobile phase with a flow rate of 0.9 ml/minute.
Pharmacokinetics analysis
The plasma time curves obtained after administration of OLM via the per-oral and sublingual routes for each animal were used to estimate pharmacokinetic parameters, and the standard deviation (SD) was calculated. Cmax (ng/ml) and Tmax (hour) were noted from the curve, which represent the maximum OLM plasma concentration (Cp) and its time, respectively. The area under the curve from zero to infinity (AUC0–∞) was calculated using the linear trapezoidal method till the Cmax was attained; after Cmax, the logarithmic method was adopted. Finally, the relative bioavailability of the sublingual route to the oral route was calculated using the following equation:
Statistical analysis
Statistical analysis and generation of the D-optimal design were completed using Design-Expert 10.0.1.0® software (Stat-Ease Inc., Minneapolis, MN), using analysis of variance (ANOVA) and regression equations. All the values are presented as mean ± SD of the mean. Comparisons between different groups were carried out using the unpaired Student’s t-test. GraphPad Prism software, version 5 (GraphPad Inc., USA), was used to carry out these statistical tests. The difference was considered to be significant at a p value < 0.05.
RESULTS AND DISCUSSION
Risk analysis
The initial step in the risk assessment study is to gather the whole information critically affecting the QTPP. This was done through a RA, which was built on defining the CQAs. In the current study, a critical attribute is the quick inclusion of the dissolution fluid. However, a conventional tablet formulation does not produce tablets with such high porosity. Thus, ODLs were prepared, owing to their highly porous structures, which allow for fast water intake, thus reducing their wetting and disintegration time (Mishra and Rohera, 2017).
The whole steps covered in the QbD within this study are summarized in Figure 1.
To facilitate risk identification, an Ishikawa diagram (Fig. 2) was constructed to represent the whole parameters affecting the studied attributes. By analyzing Figure 2, it was found that friability is considered as a key characteristic in the ODLs as it is related to the mechanical strength of the tablet, which consequently affects the ability of the ODLs to stand up to abrasion during manufacturing (Tawfeek et al., 2020). Moreover, wetting time and hence disintegration time were from the important characteristics that should be evaluated in the ODLs, to achieve fast release of the drug. Furthermore, the release rate of the drug is a crucial character of the ODLs, as it affects the bioavailability of the drug and, consequently, its pharmacological effect (Kalný et al., 2021).
A key characteristic of the ODLs is the type of the body it is formed from; thus, the type of the matrix former was studied. The effect of the four matrix formers is represented in Table 1. As can be observed, using gelatin as the matrix former resulted in a very short wetting time, with a rapid release rate, and a fast disintegration time as compared to other matrix formers. Acceptable drug content and friability % were observed with the gelatin as well and thus it could be selected as the best matrix former of the four used ones.
As shown in Table 1, the OLM content in ODLs formulations (S1–S4) was uniform and in the range of 89.113%–98.542%, which is in the acceptable range, according to the European Pharmacopoeia (EP). The EP stated that the preparation complies with the test if each content is between 85% and 115% of the average content (Van Der Steen et al., 2010). Wetting time is nearly associated with the inner structure of the tablet. The wetting time of ODLs formulations (S1–S4) was found to be in the range of 3.88–15.99 seconds. OLM-ODL S4 containing gelatin as the matrix former showed the shortest wetting time among the other formulations, which is correlated to its shortest disintegration time.
 | Figure 1. QTTP of OLM ODLs formulation. [Click here to view] |
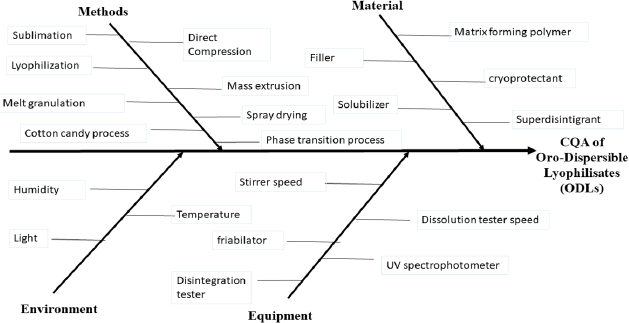 | Figure 2. Ishikawa diagram of the ODLs formulation. [Click here to view] |
The next step was the incorporation of a superdisintegrant. Recently, superdisintegrants have gained great attention due to their excellent water uptake, even at low concentrations (Mishra and Rohera, 2017). Also, a main target in this study is the enhancement of the solubility of the drug; thus, studying the effect of the superdisintegrant might have a great role.
The results of using different superdisintegrants showed that croscarmellose as the superdisintegrant is the best among the studied ones as the disintegration time of OLM-ODLs is ranked in descending order as follows: F4 (containing croscarmellose) ? F5 (containing F-melt) ? F6 (containing PEARLITOL Flash). A possible justification for these results is that both F-melt and PEARLITOL Flash are mannitol-based co-processed excipients. Therefore, their presence spontaneously increases mannitol content in the prepared formulations (Omar et al., 2017). This, in turn, leads to greater binding capacity and hydrogen bond formation between OLM and mannitol. Also, these excipients contain microcrystalline cellulose, which acquire formulations more hard core matrix that slow down disintegration time. Similar results were obtained by Jacob et al. (2007). According to them, microcrystalline cellulose and mannitol have nonwetting properties and a central rigid core, resulting in a delayed disintegration time. Croscarmellose is cross-linked cellulose acting by both swelling and wicking mechanisms; thus, it is capable of swelling 4–8-fold in less than 10 seconds (Tawfeek et al., 2020).
Croscarmellose sodium is an internally cross-linked polymer of CMC sodium. It has elevated swelling capacity and minimal gelling resulting in a rapid disintegration time. It shows a wicking action, because of its fibrous structure (Pahwa and Gupta, 2011). Thus, as stated, using croscarmellose resulted in the best results and thus will be fixed in the upcoming optimization step.
D-optimal design analysis
The D-optimal design was used as the response surface design because it allows for studying of multifactor experiments with numeric and categoric factors, where the factors can have a mixed number of levels (de Aguiar et al., 1995; Solaiman et al., 2016).
Disintegration time
Since OLM-ODLs were originated to be mainly absorbed from saliva, it is a requisite for the tablet to disintegrate in a matter of seconds and speedily dissolve. The rapid dissolution of the tablet allows the absorption of the majority of the drug in the oral cavity prior to swallowing.
As can be observed from Table 2, the disintegration time ranged from 31 to 111 seconds, which is within the acceptable limits. Although OLM-ODLs should disintegrate within 3 minutes according to the EP, there are many critics who perceive that the presence of fragments of the tablet for 3 minutes is uncomfortable for the patients, and it is favored that the tablet disintegration time be 1 minute or less (Mahmoud and Salah, 2012). Statistical analysis of the model showed a correlation coefficient of 0.9993, with a reasonable agreement between the adjusted R2 (0.9988) and predicted R2 (0.9976), and an adequate precision of 121.438, indicating the validity of the model. Further analysis of the model showed that the model was significant with significant model terms as represented in Table 3, with an insignificant lack of fit. The regression equation representing the relationship between the MA and the disintegration time is represented in the following equation:
Disintegration time = +79.25 + 11.16 * X1 + 29.97 * X2[Brij S20] − 12.81 * X2[PVP K90] − 33.21 * X2[Soluplus] − 4.69 * X1 X2[Brij S20] +5.34 * X1 X2[PVP K90] − 1.66 * X1X2 [Soluplus] − 4.80 * X12. (4)
It can be observed from Equation (4) that increasing the concentration of the solubilizer resulted in a longer disintegration time. This could be attributed to the fact that polymers at high concentrations promote the binding action of the polymer by improving wetting of the active constituents and excipient mixture inside the tablet, which leads to prolongation of the disintegration time. This observation conforms well to the results obtained previously (Shamma and Elkasabgy, 2016). The effect of the type of the solubilizer on the disintegration time is represented in Figure 3A. As could be observed, Soluplus resulted in the fastest disintegration time, which could be due to its powerful solubilizing effect, having bifunctional property, and being a strong solubilizer and a matrix former, which allows better wetting of the tablets for better disintegration (Linn et al., 2012; Shamma and Basha, 2013). The same trend was obtained by Agrawal et al. (2016).
The addition of β-cyclodextrin in preparation of OLM-ODLs resulted in the increase in the disintegration time of ODLs, which may be attributed to the poor disintegrating property of β-cyclodextrin. This seems to support the findings of Late and Banga (2010).
Cumulative amount released at 15 minutes (Q15%)
As can be observed from Table 2, the amount of the drug released within 15 minutes ranged from 65.19% to 99.42%. Statistical analysis of the model showed a correlation coefficient of 0.9839, with a reasonable agreement between the adjusted R2 (0.9737) and predicted R2 (0.9328), and an adequate precision of 33.185, indicating the validity of the model.
Further analysis of the model shows that the model was significant with significant model terms and an insignificant lack of fit, as represented in Table 3.
The regression equation representing the relationship between the MA and the amount of the drug released within 15 minutes is represented in the following equation:
Q15% = +86.23 − 5.01 * X1 − 3.68 * X2[Brij S20] + 5.31 * X2[PVP K90] + 8.32 * X2[Soluplus] + 1.34 * X1 X2[Brij S20] + 3.16 * X1 X2[PVP K90] + 0.93 * X1 X2 [Soluplus]. (5)
It can be observed from Equation (5) that there is an indirect relationship between the amount of the solubilizer and the amount of the drug released. This could be due to the increase in the binding capacity of solubilizer by increasing its concentration, which may be attributed to the amphiphilic nature of solubilizer that may hinder the penetration of dissolution medium. These findings were in accordance with Low et al. (2013), who found that the higher Soluplus-containing films exhibited slower release rates.
The effect of the type of the solubilizer on the amount of drug released rate within 15 minutes was represented in Figure 3B. As could be observed, Soluplus resulted in the greatest amount of the release of the drug, which could be due to its strong solubilizing effect that improves the solubility of BCS class II drugs (Shamma and Basha, 2013). Soluplus is usually intended for use in the aqueous instant-release film coatings formulations (Psimadas et al., 2012). Thus, Soluplus resulted in a significant reduction in the disintegration time of ODLs, which in consequence decrease the disintegration time and exhibit a faster drug release. The same trend was obtained by Agrawal et al. (2016).
The in vitro dissolution studies of all formulations were presented in Figure 4, which illustrates the remarkable effect of the addition of solubilizers in the prepared formulations on OLM release profile as the percentage of drug released from F1 to F19, in comparison to the plain drug (OLM). The percentage of drug dissolved from the plain drug after 15 minutes was only 19.30%, while the percentage of drug dissolved from different formulations was from 67.741% to 99.424%.
 | Table 3. ANOVA study of the CQAs. [Click here to view] |
 | Figure 3. Interaction plots of (A) Disintigration time, (B) amount released at 15 minutes. [Click here to view] |
Validation of the design
A design space was established where the correlation between the CPP/MA and the CQAs was defined. It enclosed the specifications of the QTPP, together with the optimized formulations. An optimized formula with the desired CQAs was suggested by the software, based on numerical optimization and desirability approach (Khafagy et al., 2020). The constraints were set to be with the least disintegration time together with the highest cumulative amount of drug release. The optimized formula (O1) as suggested by the software together with the predicted results was as represented in Table 4. O1 was formulated and tested in terms of the aforementioned CQAs, to compare these results with the expected ones (Dawoud et al., 2019). As can be observed from the small value of the % bias, the validity of the design was established. The optimized formula was then subjected to further tests.
Characterization of the optimized OLM-ODL
The drug content of the optimized OLM-ODL showed 99.08% ± 0.92%, a mean diameter of 1.4 ± 0.042 cm, and a thickness of 0.65 ± 0.065 cm. Moreover, the optimized tablets had a value of 0.176% ± 0.304% weight variation. Concerning friability studies, the percentage of weight loss of the optimized ODL was 0.308% ± 0.765%, which was in accordance with the pharmacopeial limits to be less than 1%; also, the OLM-ODLs exhibited no capping, cracking, or breakage (Basha et al., 2020). The wetting time of the optimized tablets was recorded to be 1.97 ± 0.37 seconds, which is in correlation with the disintegration time.
 | Figure 4. In-vitro drug release of OLM from ODLs containing; a) Soluplus as solubilizer, b) β-CD as solubilizer, c) PVP K90 as solubilizer, d) Brij S20 as solubilizer as well as OLM free drug and ODLs (S4) without solubilizer. [Click here to view] |
 | Table 4. Optimized OLM-ODL formula with the expected and observed results. [Click here to view] |
Physicopharmaceutical characterization of the optimized OLM-ODL
SEM analysis
SEM micrographs of the surface and cross-section views of the optimized tablet were presented in Figure 5, which showed a highly porous nature of the optimized OLM-ODL. The highly porous nature upon lyophilization elucidates the swiftness of disintegration and dissolution of OLM-ODL, providing a supreme path for water ingress and subsequent rapid wetting in the oral cavity (Kassem and Labib, 2016).
FTIR spectroscopy
Figure 6 illustrates the FTIR spectra of pure OLM, pure sucralose, and optimized OLM-ODL; changes in OLM were observed in the peaks relating to the OH functional group. These changes were manifested as broadening of the peaks. These alterations are a significant indicator of hydrogen bonding. This may be due to the formation of the cocrystal of OLM with sucralose as a potential cocrystal coformer. This process results in new crystalline structures that served to boost the drug dissolution rate compared with the parent compound (El-Gizawy et al., 2015). Similar changes have been interpreted by authors, as evidence for hydrogen bonding with the cocrystallization process (Arafa et al., 2015).
In vivo studies
Figure 7 represents the mean OLM Cp in 12 New Zealand rabbits following the administration of a drug dose equivalent to 0.25 mg/kg of OLM suspension via the per-oral route and the optimized OLM-ODL formula via the sublingual route (Of et al., 2016). The pharmacokinetic parameters for the optimized OLM-ODL formula and per-oral OLM suspension are presented in Table 5.
OLM was detected in the plasma after 5 minutes for the optimized OLM-ODL and 30 minutes for the oral route. These findings reflect more rapid absorption for the former route. The absorption rate and extent of OLM absorption were found to be different in the two routes illustrated by the higher values of Cmax by a factor of 2 and earlier Tmax for the sublingual route. The values of Cmax and Tmax for the optimized OLM-ODL were 62.2 ± 9.06 ng/ ml and 1 hour, whereas the OLM suspension values were 32.9 ± 3.71 ng/ml and 2.5 hours, respectively, which showed a significant statistical difference between the groups.
 | Figure 5. SEMs of OLM-ODL showing (a) surface view and (b) cross-section view. [Click here to view] |
 | Figure 6. IR spectrum of pure OLM, pure sucralose and the optimized OLM-ODL formula. [Click here to view] |
 | Figure 7. Cp of olmesartan via per-oral route and the optimized OLM-ODL via sublingual route. [Click here to view] |
 | Table 5. Pharmacokinetic parameters of OLM suspension via per-oral route and optimized OLM-ODL via sublingual route. [Click here to view] |
The higher value of Cmax and Tmax in the optimized OLM-ODL formula can be attributed to two main factors. The first factor is related to the formula, where rapid disintegration (Section Disintegration Time) and enhanced dissolution (Section Cumulative Amount Released at 15 minutes) were observed, and hence better absorption of the drug occurs. The second factor is correlated to the physiology of the mouth. Various physiological factors contribute to the rapid drug absorption via the sublingual route: the high vascularity of the mouth floor, where the drug is absorbed from the veins directly to the superior vena cava and bypasses the harsh GIT conditions, the first pass effect, and the thin epithelial lining of the oral mucosa.
The AUC0–24 gives insight into the amount of drug absorbed within 24 hours (Scheff et al., 2011). The AUC0–24 of the optimized OLM-ODL and OLM suspension were 365 and 106 ng.hour/ml, respectively. The optimized OLM-ODL formula showed a more than threefold higher AUC0–24 value than the OLM suspension. In addition, the relative bioavailability percentage (F) of the optimized OLM-ODL tablet was 345%, relative to the per-oral drug suspension (Table 5) (van de Donk et al., 2018). The F value proves the higher bioavailability of the optimized OLM-ODL relative to OLM suspension (Liu et al., 2012). Due to its low water solubility and efflux by drug resistance pumps in the gastrointestinal system, OLM has a low bioavailability of about 26% in humans (Lee et al., 2009). Therefore, formulating the drug with a solubilizer via the sublingual route significantly enhanced the extent of absorption and bioavailability of the drug as indicated by the AUC0–24 and F value.
CONCLUSION
The current study demonstrates a novel and comprehensive approach for the development of ODLs containing OLM, applying the QbD approach, to enhance the drug solubility, and hence its bioavailability. OLM-ODLs were prepared by the lyophilization technique. A deep product and process understanding was achieved in a risk assessment study. Formulation of the optimized formula was developed applying a D-optimal design, which studied the effects of the type and the concentration of the solubilizer on the disintegration rate and the rate of the drug release. A design space was created, and the developed optimized formula showed a rapid disintegration time of 32 seconds together with rapid release of the drug of about 6.5 mg/minute. Moreover, the optimized OLM-ODL showed promising in vivo results, when compared with the per-oral route, where the bioavailability of the OLM-ODL was almost three times higher than the per-oral route. Referring to the above findings, it could be concluded that ODLs with OLM could be successfully developed using the QbD approach, with better product and process understanding and better quality attributes.
ACKNOWLEDGMENT
The authors acknowledge Dr. Heba El-Sayed Teba, Lecturer of Organic Chemistry, for her valuable help.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
There is no funding to report.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be authors as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
REFERENCES
Abay FB, Ugurlu T. Orally disintegrating tablets: a short review. J Pharm Drug Dev, 2015; 3(3):303. CrossRef
Abd Elbary A, Ali AA, Aboud HM. Enhanced dissolution of meloxicam from orodispersible tablets prepared by different methods. Bull Fac Pharm Cairo Univ, 2012; 50(2):89–97; doi:10.1016/j.bfopcu.2012.07.001 CrossRef
Abdel Halim S. Formulation of new sildenafil citrate-caffeine orally disintegrating tablets: in vitro and in vivo evaluation. J Pharm Res Opin, 2013; 3(8):46–63.
Aghera NJ, Shah SD, Vadalia KR. Formulation and evaluation of sublingual tablets of losartan potassium. Asian Pacific J Trop Dis, 2012; 2(SUPPL.1):130–5. CrossRef
Agrawal A, Dudhedia M, Deng W, Shepard K, Zhong L, Povilaitis E, Zimny E. Development of tablet formulation of amorphous solid dispersions prepared by hot melt extrusion using quality by design approach. AAPS PharmSciTech, 2016; 17(1):214–32. CrossRef
Amasya G, Badilli U, Aksu B, Tarimci N. Quality by design case study 1: design of 5-fluorouracil loaded lipid nanoparticles by the W/O/W double emulsion—solvent evaporation method. Eur J Pharm Sci, 2016; 84:92–102. CrossRef
Arafa MF, El-Gizawy SA, Osman MA, El Maghraby GM. Sucralose as co-crystal co-former for hydrochlorothiazide: development of oral disintegrating tablets. Drug Dev Ind Pharm, 2015; 42(8):1225–33. CrossRef
Basha M, Salama A, Noshi SH. Soluplus® based solid dispersion as fast disintegrating tablets: a combined experimental approach for enhancing the dissolution and antiulcer efficacy of famotidine. Drug Dev Ind Pharm, 2020; 46(2):253–63. CrossRef
Brunner HR. Angiotensin II receptor blockers. Compr Hypertens, 2007; 2012:1003–17. CrossRef
Chauhan K, Solanki R, Sharma S. A review on fast dissolving tablet. Int J Appl Pharm, 2018; 10(6):1–7. CrossRef
Costa S de M, Lima C de A, Nobre ALCSD, Vieira D de MA, Leal ALR. Hypertension bearers with high risk/big risk of cardiovascular diseases and socioeconomic and health indicators. Rev Assoc Med Bras, 2018; 64(7):601–10. CrossRef
Danafar H, Hamidi M. LC-MS method for studying the pharmacokinetics and bioequivalence of clonidine hydrochloride in healthy male volunteers. Avicenna J Med Biotechnol, 2016; 8(2):91–8.
Dawoud MHS, Yassin GE, Ghorab DM, Morsi NM. Insulin mucoadhesive liposomal gel for wound healing: a formulation with sustained release and extended stability using quality by design approach. AAPS PharmSciTech, 2019; 20(4):158. CrossRef
de Aguiar PF, Bourguignon B, Khots MS, Massart DL, Phan-Than-Luu R. D-optimal designs. Chemom Intell Lab Syst, 1995; 30(2):199–210. CrossRef
Dey P, Ghosh A. Wafers: an innovative advancement of oro-dispersible films. Int J Appl Pharm, 2016; 8(2):16–22.
van de Donk T, Ward S, Langford R, Dahan A. Pharmacokinetics and pharmacodynamics of sublingual sufentanil for postoperative pain management. Anaesthesia, 2018; 73(2):231–7. CrossRef
El-Gendy MA, El-Assal MIA, Tadros MI, El-Gazayerly ON. Olmesartan medoxomil-loaded mixed micelles: preparation, characterization and in-vitro evaluation. Future J Pharm Sci, 2017; 3(2):90–4; doi:10.1016/j.fjps.2017.04.001 CrossRef
El-Gizawy SA, Osman MA, Arafa MF, El Maghraby GM. Aerosil as a novel co-crystal co-former for improving the dissolution rate of hydrochlorothiazide. Int J Pharm, 2015; 478(2):773–8; doi:10.1016/j.ijpharm.2014.12.037 CrossRef
El-Setouhy DA, Basalious EB, Abdelmalak NS. Bioenhanced sublingual tablet of drug with limited permeability using novel surfactant binder and microencapsulated polysorbate: in vitro/in vivo evaluation. Eur J Pharm Biopharm, 2015; 94:386–92; doi:10.1016/j.ejpb.2015.06.006 CrossRef
Fouad SA, Malaak FA, El-Nabarawi MA, Zeid KA. Development of orally disintegrating tablets containing solid dispersion of a poorly soluble drug for enhanced dissolution: in-vitro optimization/in-vivo evaluation. PLoS One, 2020; 15:1–17; doi:10.1371/journal.pone.0244646 CrossRef
Fu Y, Yang S, Jeong SH, Kimura S, Park K. Orally fast disintegrating tablets: developments, technologies, taste-masking and clinical studies. Crit Rev Ther Drug Carrier Syst, 2004; 21(6):433–75. CrossRef
Fukami J, Yonemochi E, Yoshihashi Y, Terada K. Evaluation of rapidly disintegrating tablets containing glycine and carboxymethylcellulose. Int J Pharm, 2006; 310(1–2):101–9. CrossRef
Gugulothu D, Desai P, Pandharipande P, Patravale V. Freeze drying: exploring potential in development of orodispersible tablets of sumatriptan succinate. Drug Dev Ind Pharm, 2015; 41(3):398–405. CrossRef
Hirlekar RS, Sonawane SN, Kadam VJ. Studies on the effect of water-soluble polymers on drug-cyclodextrin complex solubility. AAPS PharmSciTech, 2009; 10(3):858–63. CrossRef
Al Humaid J. Sweetener content and cariogenic potential of pediatric oral medications: a literature. Int J Health Sci (Qassim), 2018; 12(3):75–82. Available via http://www.ncbi.nlm.nih.gov/pubmed/29896075%0A; http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC5969777
ICH Expert Working Group. Pharmaceutical development Q8. In: ICH Harmonised tripartite guideline. ICH, Fukuoka, Japan, pp 1–28, 2009.
Iurian S, Bogdan C, Tomu?? I, Szabó-Révész P, Chvatal A, Leucu?a SE, Moldovan M, Ambrus R. Development of oral lyophilisates containing meloxicam nanocrystals using QbD approach. Eur J Pharm Sci, 2017; 104:356–65. CrossRef
Jacob S, Shirwaikar A, Joseph A, Srinivasan K. Novel co-processed excipients of mannitol and microcrystalline cellulose for preparing fast dissolving tablets of glipizide. Indian J Pharm Sci, 2007; 69(5):633–9. CrossRef
Kalný M, Grof Z, Št?pánek F. Microstructure based simulation of the disintegration and dissolution of immediate release pharmaceutical tablets. Powder Technol, 2021 Jan 2; 377:257–68. CrossRef
Kassem AA, Labib GS. Flash dissolving sublingual almotriptan malate lyotabs for management of migraine. Int J Pharm Pharm Sci, 2016; 9(1):125. CrossRef
Kathpalia H, Sule B, Gupte. Development and evaluation of orally disintegrating film of tramadol hydrochloride. Asian J Biomed Pharm Sci, 2013; 3(24):27–32.
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet, 2005; 365(9455):217–23. CrossRef
Khafagy ES, Fayed MH, Alrabahi SH, Gad S, Alshahrani SM, Aldawsari M. Defining design space for optimization of escitalopram ultra-fast melting tablet using suspension spray-coating technique: in-vitro and in-vivo evaluation. J Drug Deliv Sci Technol, 2020; 57:101631. CrossRef
Kumar K, Sharma PK, Srinivas L. Development of agglomerated crystals of olmesartan medoxomil by spherical crystalization technique for enhancing the micromeritic and solubility property. World J Pharm Res, 2016; 5(3):646–75.
Late SG, Banga AK. Response surface methodology to optimize novel fast disintegrating tablets using β cyclodextrin as diluent. AAPS PharmSciTech, 2010; 11(4):1627–35. CrossRef
Lee BS, Kang MJ, Choi WS, Choi YB, Kim HS, Lee SK, Lee J, Choi YW. Solubilized formulation of olmesartan medoxomil for enhancing oral bioavailability. Arch Pharm Res, 2009; 32(11):1629–35. CrossRef
Li H, Kong F, Xu J, Zhang M, Wang A, Zhang Y. Hypertension subtypes and risk of cardiovascular diseases in a Mongolian population, inner Mongolia, China. Clin Exp Hypertens, 2016; 38(1):39–44. CrossRef
Liew K Bin, Odeniyi MA, Peh KK. Application of freeze-drying technology in manufacturing orally disintegrating films. Pharm Dev Technol, 2016; 21(3):346–53. CrossRef
Linn M, Collnot EM, Djuric D, Hempel K, Fabian E, Kolter K, Lehr CM. Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur J Pharm Sci, 2012; 45(3):336–43; doi:10.1016/j.ejps.2011.11.025 CrossRef
Liu J, Shentu JZ, Wu LH, Dou J, Xu QY, Zhou HL, Wu GL, Huang MZ, Hu XJ, Chen JC. Relative bioavailability and pharmacokinetic comparison of two different enteric formulations of omeprazole. J Zhejiang Univ Sci B, 2012; 13(5):348–55. CrossRef
Low AQJ, Parmentier J, Khong YM, Chai CCE, Tun TY, Berania JE, Liu X, Gokhale R, Chan SY. Effect of type and ratio of solubilising polymer on characteristics of hot-melt extruded orodispersible films. Int J Pharm, 2013; 455(1–2):138–47. CrossRef
Mahmoud AA, Salah S. Fast relief from migraine attacks using fast-disintegrating sublingual zolmitriptan tablets. Drug Dev Ind Pharm, 2012; 38(6):762–9. CrossRef
Mishra DN, Bindal M, Singh SK, Vijaya Kumar SG. Spray dried excipient base: a novel technique for the formulation of orally disintegrating tablets. Chem Pharm Bull, 2006; 54(1):99–102. CrossRef
Mishra SM, Rohera BD. An integrated, quality by design (QbD) approach for design, development and optimization of orally disintegrating tablet formulation of carbamazepine. Pharm Dev Technol, 2017; 22(7):889–903. CrossRef
Musijowski J, Piórkowska E, Bu? - Kwa?nik K, Tycz A, Rudzki P. LC-MS method for determination of olmesartan in human plasma. PeerJ PrePrints, 2015; 3:159003. CrossRef
Narmada GY, Mohini K, Prakash Rao B, Gowrinath DXP, Kumar KS. Formulation, evaluation and optimization of fast dissolving tablets containing amlodipine besylate by sublimation method. Ars Pharm, 2009; 50(3):129–44.
Omar S, AbdAlla F, Abdelgawad N. Effect of mannitol on physical characters of lyophilized fast-disintegrating tablets. J Adv Pharm Res, 2017; 1(4):228–33. CrossRef
Pahwa R, Gupta N. Superdisintegrants in the development of orally disintegrating tablets: a review. Int J Pharm Sci Res, 2011; 2(11):2767–80.
Psimadas D, Georgoulias P, Valotassiou V, Loudos G. Molecular nanomedicine towards cancer. J Pharm Sci, 2012; 101(7):2271–80. CrossRef
Rawas-Qalaji MM, Simons FER, Simons KJ. Sublingual epinephrine tablets versus intramuscular injection of epinephrine: dose equivalence for potential treatment of anaphylaxis. J Allergy Clin Immunol, 2006; 117(2):398–403. CrossRef
Safar R, Abdelwahed W, Chehna MF, Degobert G, Fessi H. Preparation and characterization of new oral lyophilizates containing a non steroidal anti inflammatory drug. Int J Pharm Pharm Sci, 2011; 3(SUPPL. 3):108–14.
Scheff JD, Almon RR, Dubois DC, Jusko WJ, Androulakis IP. Assessment of pharmacologic area under the curve when baselines are variable. Pharm Res, 2011; 28(5):1081–9. CrossRef
Shamma R, Elkasabgy N. Design of freeze-dried Soluplus/polyvinyl alcohol-based film for the oral delivery of an insoluble drug for the pediatric use. Drug Deliv, 2016; 23(2):489–99. CrossRef
Shamma RN, Basha M. Soluplus®: a novel polymeric solubilizer for optimization of carvedilol solid dispersions: formulation design and effect of method of preparation. Powder Technol, 2013; 237:406–14; doi:10.1016/j.powtec.2012.12.038 CrossRef
Shoukri RA, Ahmed IS, Shamma RN. In vitro and in vivo evaluation of nimesulide lyophilized orally disintegrating tablets. Eur J Pharm Biopharm, 2009; 73(1):162–71; doi:10.1016/j.ejpb.2009.04.005 CrossRef
Singh H, Gupta RD, Gautam G. Formulation development, characterization, and in vitro-in vivo study of antihyperlipidemic drug rosuvastatin calcium—solid lipid nanoparticles. Asian J Pharm Clin Res, 2018 Jul; 11(7):436. CrossRef
Singh SK, Mishra DN, Jassal R, Soni P. Fast disintegrating combination tablets of omeprazole and domperidone. Asian J Pharm Clin Res, 2009; 2(4):54–62.
Solaiman A, Suliman AS, Shinde S, Naz S, Elkordy AA. Application of general multilevel factorial design with formulation of fast disintegrating tablets containing croscaremellose sodium and disintequick MCC-25. Int J Pharm, 2016; 501(1–2):87–95. CrossRef
Van Der Steen KC, Frijlink HW, Schipper CMA, Barends DM. Prediction of the ease of subdivision of scored tablets from their physical parameters. AAPS PharmSciTech, 2010; 11(1):126–32. CrossRef
Sweed NM, Fayez AM, El-Emam SZ, Dawoud MHS. Response surface optimization of self nano-emulsifying drug delivery system of rosuvastatin calcium for hepatocellular carcinoma. J Pharm Investig, 2021; 51(1):85–101; doi:10.1007/s40005-020-00497-6 CrossRef
Tashan, Emine, Karakucuk A, Celebi N. Design, and in vitro evaluation of orodispersible tablets (ODTs) of rizatriptan. AAPS PharmSciTech, 2020; 21(3):1–2. CrossRef
Tawfeek HM, Hassan YA, Aldawsari MF, Fayed MH. Enhancing the low oral bioavailability of sulpiride via fast orally disintegrating tablets: formulation, optimization and in vivo characterization. Pharmaceuticals, 2020; 13(12):446. CrossRef
Volkova TV, Simonova OR, Perlovich GL. New antifungal compound: impact of cosolvency, micellization and complexation on solubility and permeability processes. Pharmaceutics, 2021; 13(11):1865. CrossRef
Xu X, Costa AP, Khan M a., Burgess DJ. Application of quality by design to formulation and processing of protein liposomes. Int J Pharm, 2012; 434(1–2):349–59. CrossRef
Zeng Z, Chen J, Xiao C, Chen W. A global view on prevalence of hypertension and human develop index. Ann Glob Heal, 2020; 86(1):1–6. CrossRef
Zhang X, Zhang H, Ma Y, Che W, Hamblin MR. Management of hypertension using olmesartan alone or in combination. Cardiol Ther, 2017; 6(1):13–32. CrossRef